
7.6: Reparación de ADN
- Page ID
- 52986

Estrictamente definido, el mecanismo de reparación más simple no utiliza una enzima. La desalquilación o eliminación de grupos alquilo (como —CH 3 o —C2H 5) implica solo la transferencia de un grupo alquilo de una O6-metilguanina u O6-etilguanina a O6-alquilguanil-ADN alquiltransferasa. A pesar del nombre, la alquiltransferasa no es realmente una enzima, ya que está permanentemente alterada e inactivada por la reacción y por lo tanto no se ajusta a la definición de catalizador. Tenga en cuenta que esto no remedia la alquilación en N7 u otros sitios, solo los ligados a O6.
El siguiente mecanismo de reparación más simple es el desacoplamiento de los dímeros de ciclobutilo de pirimidina. Esto se puede lograr a través de la actividad de las ADN fotoliasas, también conocidas como enzimas fotoreactivadoras. Estos se denominan no solo porque la formación de los dímeros de ciclobutilo de pirimidina generalmente se debe a la exposición a la luz UV, sino porque las propias enzimas reparadoras requieren exposición a la luz (300-500 nm, cerca de UV a azul visible) para catalizar la reacción de ruptura del dímero.
Más específicamente, las ADN fotóliasas (una proteína de ~60 kD), están asociadas de manera no covalente con un cromóforo (N 5, N 10 -metileniltetrahidrofolato o 5-desazaflavina) y un FADH-. La fotoliasa se une al dímero de ciclobutilo de pirimidina de ADN monocatenario o bicatenario de una manera independiente de la luz e independiente de la secuencia. Sin embargo, no cataliza ningún cambio en el enlace hasta que la luz es absorbida por el cromóforo, que luego transfiere la energía a FADH—, provocando que se expulse y electrone al dímero, separándolo así.
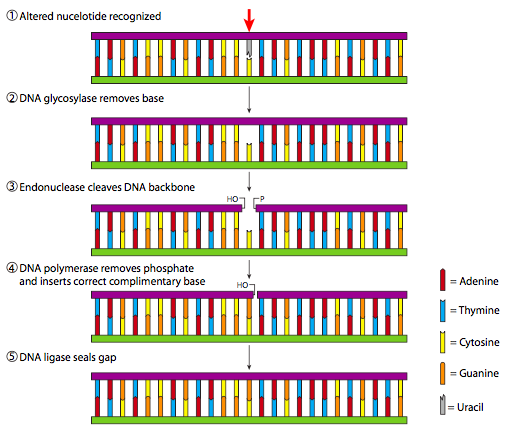
Si bien la desalquilación y la lisis de dímeros son procesos relativamente simples que hacen solo un cambio sutil en el ADN, los mecanismos de reparación por escisión son más complicados y requieren múltiples etapas enzimáticas para completarse. Cuando una lesión pequeña (no estéricamente voluminosa) se limita a una sola base, ya sea que falte en la depuración o se forme incorrectamente por desaminación o incorporación errónea, se activa el proceso conocido como reparación por escisión de base (BER). Como se ilustra en la Figura\(\PageIndex{20}\), si se reconoce una base no convencional, entonces se elimina mediante una ADN glicosilasa apropiada. En la actualidad (Genbank search, julio de 2009), existen al menos 8 genes específicos que codifican ADN glicosilasas humanas, aunque tres codifican glicosilasas que reconocen uracilo en diversas situaciones. Una vez que la base ha sido eliminada por la glicosilasa, se recluta una endonucleasa para romper los enlaces fosfodiéster que mantener la fosfodesoxirribosa ahora vacía. El hueco resultante en el ADN es rellenado por una ADN polimerasa y finalmente la cadena es reconectada por ADN ligasa.
En el caso de lesiones voluminosas que alteran significativamente la presentación física del ADN a las polimerasas y otras enzimas que procesan el ADN, se involucra un tipo diferente de proceso de reparación. La reparación de escisión de nucleótidos (NER), quizás mejor llamada reparación de escisión de poli nucleótidos, implica la eliminación de la lesión así como algunos de los nucleótidos en las inmediaciones. Hay dos iniciadores principales de la NER: o bien una porción no transcripcionalmente activa del ADN es escaneada por XPC (Figura\(\PageIndex{21}\) A), que reconoce una lesión voluminosa y recluta el complejo reparador, o cuando se transcribe un gen, la ARN polimerasa corre hacia una lesión, y luego recluta el complejo de reparación vía CSA y CSB (Figura\(\PageIndex{21}\) F y G). Si la detección es a través de XPC, uno de los factores de reparación temprana reclutados en el sitio es el Factor de Transcripción IIH/XPB/XPD, que es una helicasa de ADN (Figura\(\PageIndex{21}\) B). Este tipo de detección genómica global es ineficiente y relativamente lenta, pero proporciona un nivel basal de comprobación de errores para todo el ADN. En el caso de que el ADN se transcriba, el complejo de la ARN polimerasa ya incluye TFIIH, del que forman parte XPB y XPD. Esta detección dirigida transcripcionalmente es más eficiente y se dirige a aquellas partes del ADN de mayor uso en una célula dada. En el siguiente paso (Figura\(\PageIndex{21}\) C), XPG, asociado a BRCA1/2, y XPF, asociado a ERCC1, extirpan una porción de la hebra afectada, incluyendo pero no limitándose a la lesión misma. La ADN polimerasa δ o ε puede entonces agregar en el 3'OH libre a ll en el hueco basado en la secuencia de cadena complementaria (Figura\(\PageIndex{21}\) D). Finalmente, la reparación se conecta en su extremo 3' al resto de la cadena por ADN ligasa (Figura\(\PageIndex{21}\) E).
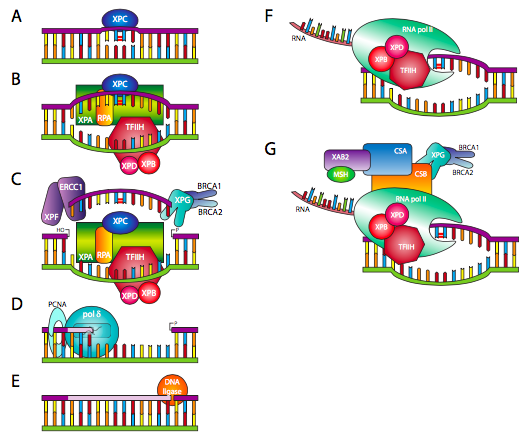
El “XP” en XPC, XPB, XPD y los demás en la Figura\(\PageIndex{21}\) se refiere a la xerodermia pigmentosa, otra enfermedad autosómica recesiva, de la cual la característica principal es la formación de carcinomas cutáneos a una edad temprana. Debido a que la NER es una forma importante de reparación de dímeros de pirimidina (además de las fotóliasas), su alteración por mutaciones en uno o más de los genes XP conduce a una sensibilidad extrema a las lesiones inducidas por UV. Los individuos afectados deben minimizar la exposición al sol. El nombre de la enfermedad proviene de las características lesiones pigmentadas (queratosis) que a menudo se forman en la piel cuando se exponen al sol.
CSA y CSB reciben el nombre del síndrome de Cockayne, un trastorno de envejecimiento autosómico recesivo. Las mutaciones en cualquiera de los genes pueden causar el trastorno, el cual se caracteriza por envejecimiento prematuro, retraso en el crecimiento, fotosensibilidad y defectos del desarrollo del sistema nervioso. Presumiblemente, noquear la capacidad de reparación del ADN de CSA o CSB conduce a una acumulación rápida de daño, incapacidad para transcribir genes necesarios y, finalmente, muerte celular.
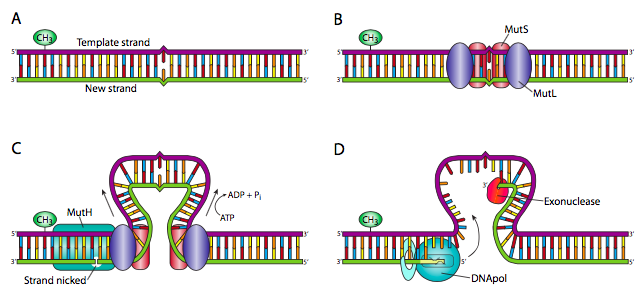
Una especie de variación en NER es el sistema de reparación de desajustes (MMR). Esto se entiende mejor en procariotas: en E. coli, MuTs es una proteína pequeña que forma homodímeros en sitios de desapareamiento. Los dímeros MuTs reclutan dos proteínas MuTl, cada una de las cuales interactúa con una de las unidades MuTs. Cada complejo Muts/Mutl empuja el ADN hacia adentro, formando un bucle con el desajuste en el centro del bucle. Esto continúa hasta que uno de los complejos Muts/mutL encuentra una secuencia GATC hemimetilada. Esto provoca el reclutamiento de MuTH, una endonucleasa altamente especializada que hace una mella monocatenaria en la cadena principal de la cadena no metilada. Esto proporciona una abertura para la exonucleasa I 3'-5' o la exonucleasa 5'-3' VII (o RecJ) para degradar la hebra desde la mella hasta el punto de desapareamiento. Esto es entonces, como habrás adivinado, rellenado por la ADN polimerasa y la columna vertebral conectada por la ligasa. En eucariotas, se han descubierto múltiples homólogos de las proteínas MuTs y MutL y el proceso es similar, pero aún no se entiende claramente, ya que aún no se ha descubierto ningún homólogo a MuTH.
Recordemos que en E. coli, las metiltransferasas de Dam finalmente metilan el ADN como método para proteger su genoma, pero el ADN recién sintetizado no está metilado. Así, la suposición es que la cadena metilada contiene la base original y correcta, mientras que el desajuste se debe a la incorporación errónea en la cadena más nueva.
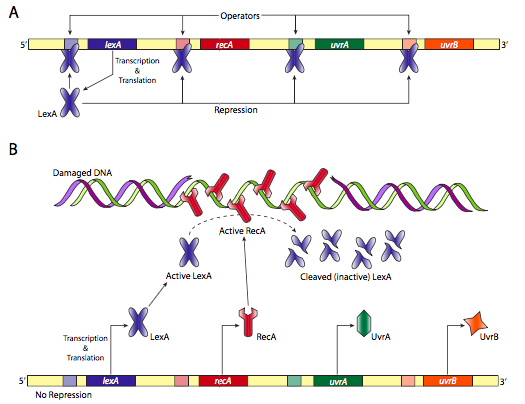
Otro sistema de reparación de ADN procariota es la respuesta SOS. Como se representa en la Figura\(\PageIndex{23}\) a continuación, si no hay daño, RecA es inactiva, por lo que la proteína LexA puede reprimir la producción de más proteínas reparadoras SOS. Sin embargo, si hay daño, las proteínas RecA se unen al ADN monocatenario y se activan. A su vez, escinden el represor LexA permitiendo la producción a partir de una serie de genes de reparación del ADN.
Hasta el momento, las reparaciones se han basado en el supuesto de que una lesión afecta solo a una hebra, y la otra hebra puede proporcionar una plantilla confiable para efectuar reparaciones sin pérdida de información. Desafortunadamente, ese no siempre es el caso, y algunas lesiones y procesos de reparación necesariamente conducen a la pérdida de secuencia. Cuando se produce una rotura bicatenaria, tal vez como producto de la radiación ionizante, el mecanismo de reparación más común se conoce como unión final no homóloga (NHEJ). Los extremos bicatenarios son reconocidos primero por Ku, una proteína circular heterodimérica que se une a los extremos del ADN. Ku luego recluta la cinasa DNA-PK CS. El DNA-PK CS actúa como un puente para unir los dos extremos, y una ADN ligasa puede unir los extremos juntos. Si las hebras se rompieron en diferentes lugares, dando como resultado voladizos monocatenarios complementarios en cada extremo (como los generados por algunas endonucleasas de restricción) entonces la reparación suele ser perfecta, ya que las secuencias complementarias alinean los dos extremos correctamente en sus posiciones originales. Sin embargo, si las nucleasas ya han actuado sobre los extremos de la cadena y ya no son complementarios, entonces la reunión de los extremos probablemente conducirá a la pérdida de información. En algunas partes del ADN, esto tendría poco efecto, pero si sucediera dentro de un gen, el producto génico mutado podría tener una función anormal o comprometida.