
11.7: Endocitosis mediada por receptores
- Page ID
- 53067
Así como hay tráfico vesicular hacia la membrana plasmática, ya sea para secreción o para incorporación de lípidos o proteínas de membrana, también puede haber tráfico vesicular desde la membrana plasmática. La endocitosis es el proceso por el cual una proteína de cubierta (generalmente clatrina) en el lado citoplásmico de la membrana plasmática, comienza a polimerizar una capa que atrae la membrana con ella hacia una vesícula. Sin embargo, en lugar de capturar un poco de ER o lumen de Golgi con él, la vesícula contiene un poco de material del exterior de la célula. A veces la endocitosis se inicia internamente, tal vez para eliminar una proteína particular de la superficie celular (por ejemplo, ver dinámica del borde posterior en la motilidad celular en el siguiente capítulo), pero a menudo, la endocitosis es el resultado de una unión de ligando a una molécula receptora extracelular, lo que lleva a su activación y posterior nucleación de un ensamblaje de clatrina y formación de vesículas.
Existen muchos tipos de ligandos: una molécula nutritiva (generalmente sobre una proteína portadora, como en los ejemplos a continuación) o incluso un virus atacante que ha cooptado por el mecanismo endecítico para facilitar la entrada a la célula. El ejemplo aquí representado es un ejemplo clásico: la endocitosis del colesterol (vía lipoproteína de baja densidad). Esto ilustra una vía potencial que pueden tomar los receptores y su carga. En el caso del colesterol, la proteína portadora se descompone por completo, aunque en el caso de la transferrina, una proteína sérica que transporta hierro en la sangre, la proteína portadora apenas se recicla después de liberar su carga de transferrina. Se empaqueta en una vesícula exocítica que se dirige de nuevo a la superficie celular.
El colesterol sérico suele estar esterificado y unido por LDL (lipoproteína de baja densidad), que luego flota en el torrente sanguíneo hasta que se encuentra con un receptor de LDL en la superficie de una célula. Cuando la LDL se une a su receptor, el receptor se activa y se forma una vesícula recubierta de clatrina alrededor del complejo LDL/receptor. Los receptores de LDL tienden a agregarse en lo que se conoce como fosas recubiertas de clatrina, vesículas parciales similares a cráteres que ya tienen un pequeño número de moléculas de clatrina polimerizadas. La vesícula se forma exactamente como se describió anteriormente para las vesículas de clatrina derivadas de Golgi: la clatrina se autoensambla en una vesícula esférica y la dinamina pellizca la vesícula de la membrana celular. Esta vesícula luego se fusiona con un endosoma temprano, que transporta bombas de protones en su membrana, haciendo que el ambiente dentro de la vesícula se acidifice (~pH 6). Esta acidificación puede provocar cambios conformacionales en las proteínas que podrían, por ejemplo, llevar a que un receptor libere su ligando, como es el caso aquí con el receptor de LDL y LDL. El endosoma temprano también funciona como una estación de clasificación: el receptor se re-vesiculariza y se transporta de regreso a la membrana plasmática. Mientras tanto, el LDL se empaqueta en una vesícula diferente y se despega para su posterior procesamiento.
Las bombas de protones endosómicas son bombas de tipo V dependientes de Mg 2 + impulsadas por ATP (a diferencia de la bomba tipo F en la membrana interna mitocondrial). Estructuralmente, los dos son similares, y la hidrólisis de ATP impulsa la unidad rotativa, que luego impulsa el movimiento de protones a través de la membrana desde el citoplasma hacia el endosoma.
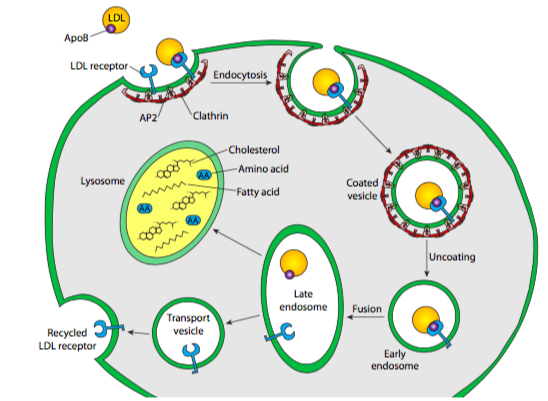
La vesícula endosómica con el LDL en ella luego se fusiona con otro compartimento ácido unido a membrana. El lisosoma, a pH ~5.0, es incluso más ácido que el endosoma, y también contiene un gran complemento de hidrolasas ácidas, enzimas hidrolíticas que van a través de sustratos (incluyendo proteasas, lipasas, glicosidasas, nucleasas) que operan óptimamente en condiciones ácidas, y mínimamente en el neutro o ligeramente condiciones básicas en el citoplasma. En parte, este es un mecanismo de seguridad —la fuga de enzimas digestivas del lisosoma no dará como resultado la digestión al por mayor de la célula porque las enzimas tienen poca o ninguna actividad en el citoplasma. La membrana lisosómica, además de tener bombas de protones para acidificar el ambiente interno, también incorpora muchas proteínas transportadoras para ayudar a mover los productos de digestión de las hidrolasas ácidas fuera del lisosoma para que la célula pueda hacer uso de los aminoácidos, azúcares, nucleótidos y lípidos que resultado. Volviendo a nuestro ejemplo, eso significa que los ésteres de colesterol se descomponen en moléculas de colesterol individuales, y la lipoproteína se descompone en lípidos y aminoácidos. Curiosamente, estas proteínas transportadoras no son digeridas por las proteasas lisosómicas porque están muy fuertemente glicosiladas, lo que protege los sitios proteolíticos potenciales de las proteasas.
Las enzimas lisosómicas son etiquetadas específicamente por una manosa-6-fosfato que se agrega en el cis Golgi. Este es un proceso de dos etapas en el que la N-acetilglucosamina fosfotransferasa agrega una fosfo-GlcNAc a un residuo de manosa, conectándose a través del grupo fosfato, luego una fosfodiesterasa elimina la GlcNAc, dejando la manosa-6-P. Esto se dirige específicamente a las enzimas lisosómicas porque todas tienen secuencias de reconocimiento de proteínas específicas a las que se une la fosfotransferasa antes de transferir la p-GlcNAc. Aunque las enzimas lisosómicas están etiquetadas en el Golgi cis, no se ordenan hasta el Golgi trans, cuando los receptores de manosa-6-P se unen a las enzimas lisosómicas y forman vesículas lisosómicas que brotarán y viajarán a endosomas tardíos y lisosomas para entregar su carga útil de hidrolasa ácida. Nuevamente, el cambio de pH es importante: en el ambiente algo ácido (pH 6.5) del Trans Golgi, el receptor se une a las enzimas etiquetadas con Manosa-6-P, pero en el lisosoma más ácido, las hidrolasas ácidas se liberan para hacer su trabajo.
Cuando una o más hidrolasas ácidas no funcionan correctamente o no llegan al lisosoma debido a una clasificación inadecuada, el resultado es una digestión incompleta del contenido lisosómico. Esto a su vez conduce a la formación de grandes inclusiones de material parcialmente digerido dentro de los lisosomas. Esta acumulación de material puede ser citotóxica, y los trastornos genéticos que afectan la expresión o clasificación de hidrolasas lisosómicas se denominan colectivamente enfermedades de almacenamiento lisosómico. Estos caen en varias categorías dependiendo de los tipos de moléculas acumuladas.
Una enfermedad común y fácilmente tratable de la acumulación de glucosaminoglicanos es la enfermedad de Hurler, que puede tratarse de manera efectiva y los efectos no neurológicos incluso revertidos por la terapia de reemplazo enzimático. Los otros de Hurler en su clase afectan a una amplia variedad de tejidos porque los glicosaminoglicanos son ubicuos. Por otro lado, debido a que el cerebro está enriquecido en gangliósidos, las enfermedades de almacenamiento lisosómico como la enfermedad de Gaucher muestran defectos principalmente en el SNC. Muchas enfermedades de almacenamiento lisosómico tienen una presentación similar: anomalías del desarrollo, especialmente retraso en el crecimiento óseo, falta de rasgos faciales finos y debilidad neuromuscular.
Dado que depende en gran medida del contenido del (de los) endosoma (s) que se fusionaron con él, el tamaño y contenido de los lisosomas pueden variar mucho. De hecho, el lisosoma también puede degradar los componentes celulares internos a través del proceso de autofagia. Por lo general, esto se inicia bajo condiciones de inanición que conducen a la inhibición de mTOR, y posterior expresión de genes autofágicos. Estos luego interactúan con las mitocondrias y otros componentes celulares, y promueven la formación de un autofagosoma de doble membrana a su alrededor. El origen de las membranas no está claro, aunque se sospecha la ER. Finalmente, el autofagosoma se fusiona con un lisosoma, y las hidrolasas ácidas descomponen las partes celulares para obtener energía. También puede ocurrir una variación de esta llamada microautofagia, en la que el lisosoma mismo invagina un poco de material citoplásmico e internaliza una vesícula intralisosómica que luego se descompone.
La enfermedad de células I más grave (mucolipidosis tipo II) ocurre cuando casi todas las enzimas lisosómicas faltan en los fibroblastos del individuo afectado. Hay retraso grave en el desarrollo y falla temprana del crecimiento, problemas neuromusculares y malformaciones en el desarrollo esquelético temprano. La gravedad de este trastorno se debe a la falta casi completa de enzimas lisosómicas, la cual es causada por una deficiencia de GlcNAc fosfotransferasa. Sin ella, no se etiquetan enzimas para clasificar al lisosoma.
Otros trastornos relativamente comunes incluyen las enfermedades Tay-Sachs y Niemann-Pick. Tay-Sachs es causado por una acumulación de gangliósidos en el cerebro y suele ser fatal a los 5 años de edad. Niemann-Pick, en cambio, puede manifestarse como Tipo A con una esperanza de vida aún más corta, o como Tipo B, en el que los síntomas son relativamente menores. La principal diferencia es que los pacientes de Tipo A tienen muy poca (< 5%) de su actividad de esfingomielinasa, mientras que los pacientes de Tipo B tienen solo una actividad ligeramente inferior a la normal (~ 90%).
Por último, cabe señalar que las grandes vacuolas de células vegetales son de hecho lisosomas especializados. Recordemos que las vacuolas ayudan a mantener la turgencia, o presión de agua hacia afuera sobre las paredes celulares que conducen a una parte rígida de la planta en lugar de una flácida, marchita. Una de las formas en que esto ocurre es que las hidrolasas ácidas dentro de la vacuola alteran la presión osmótica dentro de la vacuola para regular el movimiento del agua ya sea dentro o fuera.
Otro ejemplo de endocitosis mediada por receptores es la importación de hierro a una célula de mamífero. Al igual que con el colesterol sérico, el hierro generalmente no se importa a la célula por sí mismo. En cambio, se une a la apotransferrina, una proteína sérica que une dos iones Fe 3 +. Una vez que se ha unido a los iones de hierro, la apotransferrina se conoce ahora como transferrina, y puede ser reconocida y unida por receptores de transferrina (TfR) ubicados en la superficie extracelular de las membranas celulares. Esto inicia la endocitosis mediada por receptores tal como se describió anteriormente. Sin embargo, en este caso, el lisosoma no está involucrado. A medida que la transferrina y el receptor de transferrina alcanzan el endosoma temprano, no se disocian, sino que el Fe 2 + libera de la transferrina, y luego sale del endosoma vía DMT1, una proteína de transporte de metal divalente para ser utilizada en grupos hemo u otros complejos. Esto deja el complejo apotransferrina-TFR, que se recicla de nuevo a la membrana celular a través de vesículas. Una vez que la vesícula se fusiona con el espacio extracelular, la acidez del endosoma se disipa y la apotransferrina ya no se une a TfR. La apotransferrina puede así volver a su deber de encontrar iones de hierro y devolverlos a la célula.