
12.8: Dinámica citoesquelética
- Page ID
- 53334
En el desarrollo temprano de los animales, hay una gran cantidad de reordenamiento celular y migración ya que la gota aproximadamente esférica de células llamada blástula comienza a diferenciarse y formar células y tejidos con funciones especializadas. Estas células necesitan pasar de su punto de nacimiento a sus eventuales posiciones en el animal completamente desarrollado. Algunas células, como las neuronas, tienen un tipo adicional de motilidad celular: extienden procesos largos (axones) desde el cuerpo celular hasta su objetivo de inervación. Tanto en la extensión de neuritas como en la motilidad celular completa, la célula necesita mover primero sus puntos de unión y luego el grueso de la célula de un punto a otro. Esto se hace gradualmente, y utiliza el citoesqueleto para hacer más eficiente el proceso. Los principales elementos en la motilidad celular son el cambio del punto de adhesión hacia adelante, la limpieza del espacio interno mediante el reordenamiento de microfilamentos de actina con miosina y el posterior llenado de ese espacio con microtúbulos.
Para que la fuerza se transmita, la membrana debe estar unida al citoesqueleto. De hecho, la señalización de los receptores en la membrana a veces puede inducir directamente reordenamientos o movimientos del citoesqueleto a través de proteínas adaptadoras que conectan la actina (u otros elementos citoesqueléticos) con proteínas transmembrana como los receptores de integrina. Uno de los primeros sistemas experimentales para estudios de interacción citoesqueleto-membrana fue el eritrocito (glóbulo rojo).
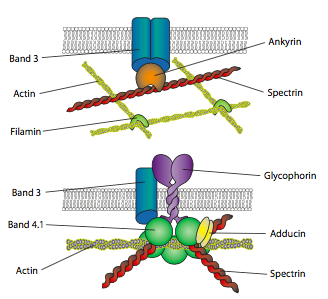
Las ilustraciones anteriores (Figura\(\PageIndex{15}\)) muestran algunas de las interacciones de una extensa red de microfilamentos de actina con proteínas transmembrana. La anquirina y la espectrina son importantes proteínas de enlace entre las proteínas transmembrana y los microfilamentos. Esta idea de construir un complejo proteico alrededor del lado citoplasmático de una proteína transmembrana es ubicua, y las proteínas de andamiaje (enlace) se utilizan no solo para conectar el sustrato extracelular (vía proteína transmembrana) al citoesqueleto, sino también para conectar físicamente moléculas de señalización y así aumentar la velocidad y eficiencia de la transducción de señales.
Las proteínas accesorias a actinfilamentos y microtúbulos se mencionaron brevemente antes. Entre otras funciones, pueden controlar la polimerización y despolimerización, formar haces, organizar redes y tender puentes entre las diferentes redes citoesqueléticas. Para la actina, las proteínas primarias de control de la polimerización son la profilina, que promueve la polimerización y la timosina β 4, que secuestra la g-actina. Las proteínas de protección final menos complejo Arp 2/3 y tropomodulina, y las proteínas capZ, severina y gelsolina pueden estabilizar los extremos de la f-actina. Finalmente, la cofilina puede aumentar la despolimerización desde el extremo (-).
La profilina tiene dos actividades que promueven la polimerización. Primero, es un factor de intercambio de nucleótidos que elimina el ATP unido a la g-actina, y lo reemplaza con ADP. Esto suena contradictorio, pero sigue leyendo hasta el siguiente párrafo. Segundo, cuando se une a una g-actina, aumenta la tasa de adición a los microfilamentos de actina. Lo hace uniéndose al extremo opuesto al sitio de unión a ATP, dejando ese sitio y ese lado abiertos para unirse tanto a ATP como al extremo (+) de un microfilamento. La profilina se puede encontrar tanto en el citoplasma en general como asociada con fosfolípidos (PIP 2) y proteínas de membrana, para controlar procesos tales como la remodelación del borde de ataque de las estructuras citoesqueléticas de f-actina.
La timosina β 4 regula el ensamblaje de microfilamentos controlando el charco disponible de g-actina. Ya afirmamos que mayores concentraciones de g-actina pueden aumentar las tasas de polimerización. Sin embargo, debido a la naturaleza altamente dinámica del citoesqueleto de actina, las limitaciones de tiempo de degradar y producir nueva actina evitarían el control de respuesta rápida necesario. Por lo tanto, el mecanismo óptimo es mantener un gran acervo de monómeros de g-actina, pero regular su disponibilidad atándolo con una proteína secuestrante, la timosina β 4. La timosina β 4 tiene una afinidad 50 veces mayor por G-actina-ATP que por G-actina-ADP, así que aquí es donde la profilina vuelve a aparecer. Profilina intercambia el ATP de un complejo Tβ 4-G-actina-ATP por un ADP. El resultado es que el Tβ 4 libera el G-actina-ADP, lo que le permite ingresar a la piscina general para construir filamentos.
El aumento de la despolimerización y la ralentización o el cese de la polimerización pueden descomponer gradualmente las estructuras de f-actina, pero ¿y si hay una necesidad de una descomposición rápida? Dos de las proteínas de capoteo mencionadas anteriormente, gelsolina y severina, tienen un modo de acción alternativo que puede cortar microfilamentos de actina en cualquier punto uniéndose junto a un filamento de actina y alterando la conformación de la subunidad a la que se une. El cambio conformacional obliga a la interacción actina-actina a romperse, y la gelsolina o severina permanece en su lugar como una proteína de protección terminal (+).
La gelsolina es inhibida por el fosfolípido PIP 2. La fosfolipasa C, que descompone la PIP 2 también puede aumentar el Ca 2+ citosólico, que es un activador de la gelsolina. Por lo tanto, es posible incrementar rápidamente la actividad de gelsolina mediante señalización PLC.
En el lado de los microtúbulos, debido a la inestabilidad dinámica, se podría pensar que no se necesita una enzima seccionadora, pero de hecho, la espastina y la katanina son proteínas de corte de microtúbulos que se encuentran en una variedad de tipos celulares, particularmente neuronas. También existe una proteína similar a Tβ 4 para la tubulina: Op18, o estathmin, que se une a dímeros de tubulina (no monómeros), actuando para secuestrarlos y disminuir la concentración de trabajo. Está regulado por la fosforilación (que desactiva su unión a tubulina).
Las mutaciones en la espastina están ligadas al 40% de aquellas paraplejias espásticas que se distinguen por la degeneración de axones muy largos. La capacidad de corte de la espastina parece ser necesaria para la remodelación del citoesqueleto en respuesta al daño neuronal.
Las proteínas MAP1, MAP2 y tau (t) asociadas a microtúbulos trabajan para promover el ensamblaje de microtúbulos, así como otras funciones. MAP1 es el más generalmente distribuido de los tres, con tau que se encuentra principalmente en las neuronas, y MAP2 aún más restringido a las dendritas neuronales. Estos y algunos otros MAP también actúan para estabilizar los microtúbulos frente a catástrofes al unirse junto al microtúbulo y reforzar las interacciones tubulina-tubulina.
Tau tiene una historia biomédica complicada. Su función normal es clara: ensamblar, estabilizar y unir microtúbulos. Sin embargo, también se encuentra en ovillos neurofibrilares hiperfosforilados que se asocian con la enfermedad de Alzheimer. Aún no se conoce una causa de Alzheimer, por lo que aún no está claro si los enredos de la proteína tau juegan un papel importante en alguno de los síntomas.
Finalmente, con respecto a las proteínas accesorias de microfilamentos y microtúbulos, están los enlazadores. Algunos de los MAP mencionados anteriormente pueden reticular microtúbulos en matrices paralelas o de malla, al igual que algunas cinesinas y dineinas, aunque convencionalmente se consideran proteínas motoras. En el lado de los microfilamentos, hay muchas proteínas conocidas que reticulan la f-actina, muchas de las cuales están en la superfamilia del dominio de homología de calponina, incluyendo fimbrina, α-actinina, β-espectrina, distrofina y filamina. Aunque todas pueden unirse a la actina, la forma de la proteína dicta diferentes tipos de interacción: por ejemplo, la fimbrina principalmente agrupa la f-actina en paralelo para formar haces, mientras que la filamina une los filamentos de actina perpendicularmente para formar redes de malla.
El síndrome FG es una enfermedad genéticamente ligada que se caracteriza por retraso mental, agrandamiento de la cabeza, hipotonía congénita, ano imperforado y agenesia parcial del cuerpo calloso. Se ha vinculado a mutaciones en varios genes del cromosoma X, entre ellos la filamina A (FLNA, FLN1, localizada Xq28).
Las mutaciones en la distrofina, que es una proteína muscular importante de la superfamilia del dominio CD, pueden resultar en Distrofia Muscular de Duchenne o la Distrofia Muscular Becker relacionada pero menos severa. La característica más distintiva es una degeneración muscular proximal progresiva y pseudohipertrofia de los músculos de la pantorrilla. El inicio de la DMD generalmente se reconoce antes de los 3 años y es letal a los 20 años. Sin embargo, los síntomas de DMO pueden no presentarse hasta los años 20, con buena probabilidad de supervivencia a largo plazo. Aunque es principalmente una enfermedad de desgaste muscular, la distrofina está presente en otros tipos de células, incluidas las neuronas, lo que puede explicar un vínculo con el retraso mental leve en algunos pacientes con DMD. Al igual que FLNA, el gen de la distrofina también se localiza en el cromosoma X (Xp21.2).