
14.1: Antecedentes
- Page ID
- 53863

La SDS-PAGE es ampliamente utilizada para analizar las proteínas en extractos complejos. Los métodos más utilizados se derivan del sistema SDS-PAGE discontinuo descrito por primera vez por Laemmli (1970). El sistema en realidad consiste en dos geles: un gel de resolución (también conocido como correr) en el que las proteínas se resuelven en función de sus pesos moleculares (MW) y un gel apilable en el que las proteínas se concentran antes de entrar en el gel de resolución. Las diferencias en las composiciones del gel apilable, el gel de resolución y el tampón de electroforesis producen un sistema que es capaz de resolver finamente proteínas de acuerdo con sus MW.
Electroforesis en gel de macromoléculas
En la electroforesis en gel, se utiliza un campo eléctrico para mover moléculas cargadas a través de
una matriz de una sustancia polimerizada como agarosa o poliacrilamida. Las velocidades a las que las moléculas individuales se mueven a través del gel dependen de las propiedades tanto del sistema de separación como de las propias moléculas. Las matrices de gel están permeadas con redes de poros a través de las cuales se mueven las moléculas. La cantidad de resistencia que presenta la matriz al movimiento de una molécula depende del diámetro del poro así como del tamaño y geometría de la molécula. Los investigadores pueden controlar el tamaño del poro ajustando la concentración de monómero de gel dentro de un cierto rango. En general, las moléculas más pequeñas y con mayor carga migran más rápidamente a través de geles que las moléculas más grandes o menos cargadas. La movilidad de una molécula también se ve afectada por el sistema tampón y la fuerza del campo electroforético utilizado para la separación.
Ya has usado electroforesis en gel de agarosa para separar moléculas de ADN. Recordemos que el tamaño de una molécula de ADN lineal puede estimarse a partir de la velocidad a la que se mueve a través de un gel de agarosa, debido a que las moléculas de ADN tienen una relación carga a masa uniforme. La electroforesis de proteínas es algo más complicada que la electroforesis de ADN. Las proteínas son mucho más pequeñas que las moléculas de ADN, por lo que se utilizan geles de poliacrilamida para su separación. Además, las proteínas son mucho más estructuralmente diversas que el ADN, por lo que se utilizan tratamientos químicos (ver más abajo) para impartir una geometría uniforme y una relación carga/masa a las proteínas.
Química de la polimerización de acrilamida
Los geles de poliacrilamida utilizados para separar proteínas están formados por la polimerización química de acrilamida y un reactivo de reticulación, N, N'metilenbisacrilamida (página opuesta). Los investigadores son capaces de controlar el tamaño de los poros en el gel ajustando la concentración de acrilamida, así como la relación de acrilamida a bisacrilamida. Elevar la concentración de acrilamida o bisacrilamida, mientras se mantiene constante la otra concentración, disminuirá el tamaño de poro del gel. La polimerización se produce debido a los radicales libres de oxígeno que reaccionan con los grupos vinilo en acrilamida y bisacrilamida, como se muestra en la siguiente figura. Los radicales oxígeno se generan a partir del catalizador, persulfato amónico (APS), cuando reacciona con un segundo catalizador, N, N', N'-tetrametiletilendiamina (TEMED).
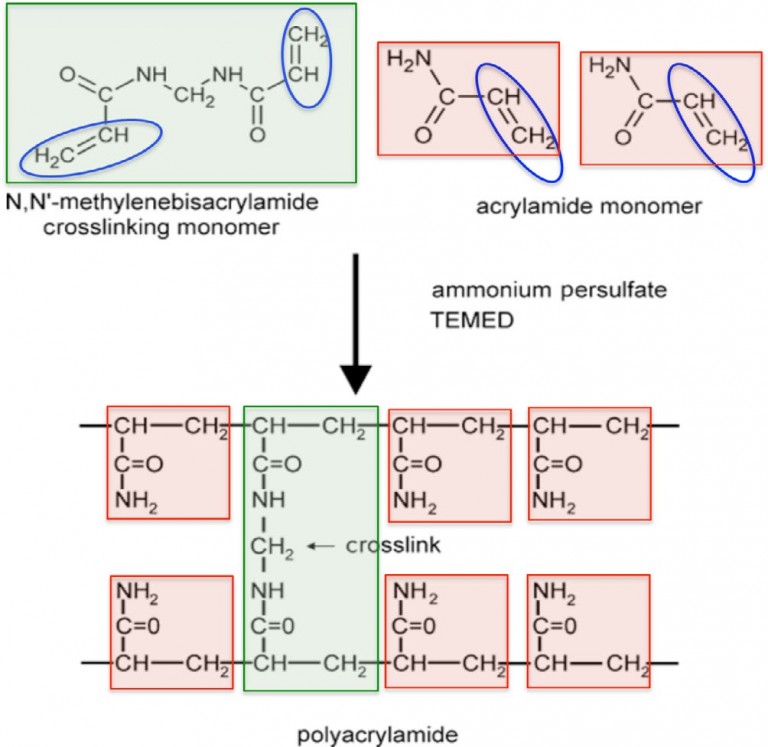
Polimerización en gel de
El persulfato de amonio y TEMED catalizan la polimerización de monómeros de acrilamida y bis-acrilamida en una red reticulada.
Las proteínas se desnaturalizan antes de la electroforesis
En comparación con las moléculas de ADN, las proteínas son estructuralmente muy diversas. Las proteínas muestran una tremenda variación en sus composiciones de aminoácidos y en la distribución de aminoácidos
en sus estructuras plegadas, características con importantes implicaciones para la electroforesis. Recordemos que las proteínas son mezclas de aminoácidos hidrófobos e hidrófilos y que la secuencia primaria de la proteína determina su forma plegada final. Debido al efecto hidrofóbico, las superficies
de proteínas proteínas tienen una mayor frecuencia de aminoácidos polares y cargados que los interiores, donde predominan los residuos hidrófobos. Las proteínas plegadas asumen muchas geometrías diferentes y sus superficies son mosaicos con respecto a la distribución de grupos R con diferentes químicas. Debido a que las proteínas son tan diversas con respecto a sus cargas superficiales y geometrías, los pesos moleculares de las proteínas plegadas no pueden determinarse simplemente por su tasa de migración en un campo eléctrico. ¡Las proteínas cargadas postiva y negativamente migrarían en diferentes direcciones!
Para resolver las proteínas en una muestra de acuerdo a su tamaño, los investigadores deben convertir las proteínas a una geometría uniforme e impartir una relación carga/masa uniforme a las proteínas. En SDS-PAGE, la solución consiste en desnaturalizar las proteínas hirviéndolas con el detergente aniónico, dodecilsulfato de sodio (SDS) y 2-mercaptoetanol. La combinación de calor y detergente es suficiente para romper los muchos enlaces no covalentes que estabilizan los pliegues proteicos, y el 2-mercaptoetanol rompe cualquier enlace covalente entre los residuos de cisteína. Al igual que otros detergentes, el SDS es una molécula anfipática, que consiste en una cadena hidrofóbica de 12 carbonos y un grupo sulfato hidrófilo. La cadena hidrocarbonada SDS impregna el interior de la proteína y se une a grupos hidrófobos, reduciendo la proteína a una espiral aleatoria, recubierta con moléculas de detergente cargadas negativamente a lo largo de su longitud. Las proteínas desnaturalizadas se unen a una gran cantidad de SDS, lo que equivale a ~1.4 g de SDS/g de proteína, o ~una molécula de SDS por cada dos aminoácidos.
Las discontinuidades entre los geles de apilamiento y de ejecución subyacen al poder de resolución de los geles SDS-PAGE
El sistema Laemmli (1970) SDS-PAGE puede ser considerado como un sistema de 3 componentes. Los geles de apilamiento y ejecución (resolución) tienen diferentes tamaños de poro, fuerzas iónicas y pH diferentes. El tercer componente es el tampón de electroforesis (Tris 25 mM, glicina 192 mM, SDS 0.1%, pH ~8.3), que contiene grandes cantidades de glicina. El estado de ionización de la glicina es crítico para la separación. A pH neutro, la glicina es un zwitterión, con un grupo carboxilo cargado negativamente y un grupo amino cargado positivamente. El pK a del grupo amino es 9.6, considerablemente mayor que el pH del tampón de cámara. En consecuencia, muy poca glicina tiene una carga negativa en el tampón de la cámara o gel de apilamiento, y no se produce una ionización significativa hasta que la glicina entra en el ambiente de pH 8.8 más alcalino del gel en funcionamiento. Sigamos el progreso de las muestras de proteínas durante la SDS-PAGE para ver cómo las diferencias en la composición de estos tres componentes generan el alto poder de resolución de los geles SDS-PAGE.
como un sistema de 3 componentes. Los geles de apilamiento y ejecución (resolución) tienen diferentes tamaños de poro, fuerzas iónicas y pH diferentes. El tercer componente es el tampón de electroforesis (Tris 25 mM, glicina 192 mM, SDS 0.1%, pH ~8.3), que contiene grandes cantidades de glicina. El estado de ionización de la glicina es crítico para la separación. A pH neutro, la glicina es un zwitterión, con un grupo carboxilo cargado negativamente y un grupo amino cargado positivamente. El pK a del grupo amino es 9.6, considerablemente mayor que el pH del tampón de cámara. En consecuencia, muy poca glicina tiene una carga negativa en el tampón de la cámara o gel de apilamiento, y no se produce una ionización significativa hasta que la glicina entra en el ambiente de pH 8.8 más alcalino del gel en funcionamiento. Sigamos el progreso de las muestras de proteínas durante la SDS-PAGE para ver cómo las diferencias en la composición de estos tres componentes generan el alto poder de resolución de los geles SDS-PAGE.
El tampón de muestra utilizado para SDS-PAGE contiene un colorante de rastreo, azul de bromofenol (BPB), que migrará con el borde de ataque de las proteínas separadas en el gel. El tampón de muestra Th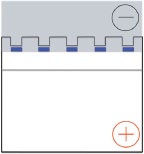 e también contiene glicerol, lo que permite que las muestras de proteína se asienten en el fondo de los pocillos de gel. El gel se coloca verticalmente en el aparato de electroforesis y se cubre con tampón de cámara que contiene glicina (derecha, sombreada).
e también contiene glicerol, lo que permite que las muestras de proteína se asienten en el fondo de los pocillos de gel. El gel se coloca verticalmente en el aparato de electroforesis y se cubre con tampón de cámara que contiene glicina (derecha, sombreada).
Una vez que se aplica un voltaje, los iones cloruro en el tampón de muestra y el gel de apilamiento se mueven rápidamente hacia el polo positivo, formando el borde de ataque de un frente de iones en movimiento. 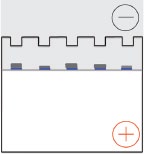 Las moléculas de glicina tienen muy poca carga en el gel apilable, por lo que migran en la parte posterior del frente de iones móviles. Esta diferencia en la movilidad de cloruro y glicina establece un gradiente de voltaje pronunciado en el gel de apilamiento que barre a lo largo de los complejos proteína-SDS cargados negativamente. Los poros grandes del gel apilable presentan muy poca resistencia al movimiento de los complejos proteína-SDS, que luego se “apilan” en una región muy concentrada en la interfaz entre los geles de ejecución y apilamiento (derecha). Los complejos proteína-SDS permanecen concentrados en la interfaz hasta que las moléculas de glicina que migran lentamente alcanzan el límite entre los dos geles.
Las moléculas de glicina tienen muy poca carga en el gel apilable, por lo que migran en la parte posterior del frente de iones móviles. Esta diferencia en la movilidad de cloruro y glicina establece un gradiente de voltaje pronunciado en el gel de apilamiento que barre a lo largo de los complejos proteína-SDS cargados negativamente. Los poros grandes del gel apilable presentan muy poca resistencia al movimiento de los complejos proteína-SDS, que luego se “apilan” en una región muy concentrada en la interfaz entre los geles de ejecución y apilamiento (derecha). Los complejos proteína-SDS permanecen concentrados en la interfaz hasta que las moléculas de glicina que migran lentamente alcanzan el límite entre los dos geles.
Se producen cambios dramáticos a medida que los iones glicina entran en el gel en funcionamiento. El pH del gel en marcha está más cerca del pKa de los grupos amino de glicina, por lo que una fracción significativa de las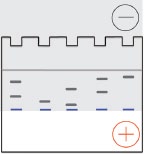 moléculas de glicina asumen una carga negativa. Las moléculas de glicina cargadas negativamente comienzan a moverse a la misma velocidad que los iones cloruro, eliminando así la diferencia de voltaje que controlaba la movilidad de las proteínas a través del gel de apilamiento. Los poros en el gel circulante son mucho más pequeños que los del gel apilable, por lo que los poros presentan resistencia friccional a la migración de proteínas. Las proteínas comienzan a migrar a diferentes velocidades, debido a las propiedades de tamizado del gel. Los complejos proteína-SDS más pequeños migran más rápidamente que los complejos proteína-SDS más grandes (derecha). Dentro de un cierto rango determinado por la porosidad del gel, la tasa de migración de una proteína en el gel en marcha es inversamente proporcional al logaritmo de su PM.
moléculas de glicina asumen una carga negativa. Las moléculas de glicina cargadas negativamente comienzan a moverse a la misma velocidad que los iones cloruro, eliminando así la diferencia de voltaje que controlaba la movilidad de las proteínas a través del gel de apilamiento. Los poros en el gel circulante son mucho más pequeños que los del gel apilable, por lo que los poros presentan resistencia friccional a la migración de proteínas. Las proteínas comienzan a migrar a diferentes velocidades, debido a las propiedades de tamizado del gel. Los complejos proteína-SDS más pequeños migran más rápidamente que los complejos proteína-SDS más grandes (derecha). Dentro de un cierto rango determinado por la porosidad del gel, la tasa de migración de una proteína en el gel en marcha es inversamente proporcional al logaritmo de su PM.
Las proteínas se visualizan con manchas.
Con pocas excepciones, las proteínas naturales son invisibles en los geles SDS-PAGE. En consecuencia, los investigadores a menudo usan estándares de proteínas teñidas previamente para monitorear las posiciones aproximadas de las proteínas durante la electroforesis. Los patrones pre-teñidos se producen uniendo covalentemente un gran número de cromóforos a una proteína. La adición de los cromóforos aumenta el PM de la proteína y también produce bandas más difusas en el
gel. La difusividad de las bandas refleja la variación en el número de moléculas de colorante unidas a moléculas proteicas individuales. Usaremos escaleras estándar de proteínas preteñidas en nuestros geles, para que pueda visualizar la separación de proteínas que se está produciendo. Las proteínas de levadura no serán visibles, sin embargo, porque no se han modificado con cromóforos.
Para visualizar las posiciones de las proteínas una vez completada la electroforesis, los investigadores tiñen los geles con diversos colorantes que se unen de manera no covalente y con muy poca especificidad a las proteínas. Durante el proceso de tinción, las proteínas también se “fijan” en el gel, lo que significa que las proteínas se vuelven insolubles e incapaces de difundirse fuera del gel. En nuestros experimentos, utilizaremos Simply Blue, una suspensión coloidal de Coomassie Brilliant Blue G-250. El Azul Brillante G-250 se une a proteínas de forma no específica a través de un gran número de interacciones iónicas y Van der Waals. En este procedimiento, los geles se enjuagan con agua para eliminar las sales tampón utilizadas para la electroforesis y luego se tratan con la suspensión coloidal G-250. Las bandas de proteínas aparecen rápidamente, y cuando es necesario, los geles pueden ser desteñidos con agua desionizada para disminuir el fondo del gel. La intensidad de tinción con Azul Brillante se considera un procedimiento cuantitativo, ya que con algunas excepciones, la intensidad de una banda teñida es directamente proporcional a la cantidad de proteína en una banda.
Los pesos moleculares de las proteínas se pueden calcular a partir de su migración en geles
Los tamaños de las proteínas en un extracto se pueden calcular comparando su migración con un conjunto de proteínas estándar ejecutadas en el mismo gel. Los investigadores seleccionan proteínas estándar que se resolverán bien en el gel particular que están ejecutando. Por ejemplo, un investigador que utilice un gel de 7.5% seleccionará estándares con pesos moleculares más altos (MW) que un investigador que utilice un gel al 15%, el cual es más adecuado para el análisis de proteínas pequeñas. Los principios utilizados para estimar MW son los mismos utilizados para la electroforesis en gel de agarosa. Una gráfica del log 10 MW de las proteínas estándar frente a la distancia que migró cada proteína sobre el gel dará una línea recta en la región donde el gel tiene buen poder de resolución. (Nota: MW no es lo mismo que la masa de una proteína. MW es un término adimensional. Por ejemplo, la mioglobina tiene una masa de 16.7 kDa y un MW de 16.700.) Los tamaños de proteínas desconocidas se pueden estimar interpolando valores experimentales en una gráfica de proteínas estándar. Las proteínas cuyos pesos moleculares se encuentran fuera de este rango no se resolverán bien en el gel.
La siguiente figura ilustra varios de los puntos discutidos anteriormente. Los mismos conjuntos de patrones de proteína no teñidos y pre-teñidos se separaron en geles SDS-PAGE 12% o 15%. Los patrones preteñidos en los carriles 1-5 son visibles sin tinción, pero se vuelven mucho más pronunciados después de la tinción. El patrón no teñido en el carril 6 requiere tinción para hacerse visible, pero las bandas son mucho más discretas y darán valores más confiables al calcular MW de proteínas desconocidas, debido a que los cromóforos no se han unido a las proteínas. Los datos en los carriles 2-5 también demuestran que la tinción con Azul Brillante es un procedimiento cuantitativo, debido a que la intensidad de las bandas en cada carril aumenta en proporción directa a la cantidad de proteína en el carril.
Al analizar sus datos experimentales, recuerde considerar los aminoácidos adicionales que se han agregado durante el procedimiento de clonación. Las proteínas Met con las que está trabajando son proteínas de fusión con aminoácidos adicionales en los extremos C y las proteínas Met. El plásmido BG1805 codifica epítopos HA e His6, así como el dominio de unión a inmunoglobulina ZZ. Juntas, estas secuencias añaden un valope de ~19 kDa a la masa esperada de las proteínas Met de S. cerevisiae (Gelperin et al., 2005). El plásmido pYES2.1 codifica 33 aminoácidos que se añaden a los ORF clonados. Las secuencias adicionales incluyen una etiqueta de epítopo V5 y una etiqueta de purificación (His) 6 en los extremos C de proteínas sobreexpresadas. Juntos, estos aminoácidos agregan ~5 kDa al tamaño de la proteína.
El PM de la proteína de control LacZ sin el epítopo V5 es de ~120 kDa. Debido a que esta es una proteína tan grande, será muy difícil obtener una estimación precisa de su MW.