
17.2: Introducción a los motivos reguladores y la regulación génica
- Page ID
- 54141

Ya hemos explorado las áreas de programación dinámica, alineación de secuencias, clasificación y modelado de secuencias, modelos ocultos de Markov y maximización de expectativas. En el siguiente capítulo, veremos cómo estas técnicas también son útiles para identificar nuevos motivos y dilucidar sus funciones.
El código regulatorio: Factores y Motivos de Transcripción
Los motivos son cortos (6-8 bases de largo), patrones recurrentes que tienen funciones biológicas bien definidas. Los motivos incluyen patrones de ADN en regiones potenciadoras o motivos promotores, así como motivos en secuencias de ARN tales como señales de corte y empalme. Como hemos comentado, la actividad genética está regulada en respuesta a las variaciones ambientales. Los motivos son responsables de reclutar Factores de Transcripción, o proteínas reguladoras, para el gen diana apropiado. Los motivos también pueden ser reconocidos por microARN, que se unen a motivos dados a través de la complementariedad; nucleosomas, que reconocen motivos basados en su contenido de GC; y otros ARN, que utilizan una combinación de secuencia y estructura de ADN. Una vez unidos, pueden activar o reprimir la expresión del gen asociado.
Los factores de transcripción (TFs) pueden utilizar varios mecanismos para controlar la expresión génica, incluyendo la acetilación y desacetilación de proteínas histonas, el reclutamiento de moléculas de cofactor en el complejo TF-ADN y la estabilización o alteración de las interfaces ARN-ADN durante la transcripción. A menudo regulan un grupo de genes que están involucrados en procesos celulares similares. Por lo tanto, es probable que los genes que contienen el mismo motivo en sus regiones aguas arriba estén relacionados en sus funciones. De hecho, muchos motivos reguladores se identifican analizando las regiones aguas arriba de genes que se sabe que tienen funciones similares.
Los motivos se han vuelto extremadamente útiles para definir redes reguladoras genéticas y descifrar las funciones de los genes individuales. Con nuestras habilidades computacionales actuales, el descubrimiento y análisis de motivos reguladores ha progresado considerablemente y se mantiene a la vanguardia de los estudios genómicos.
Desafíos del descubrimiento de motivos
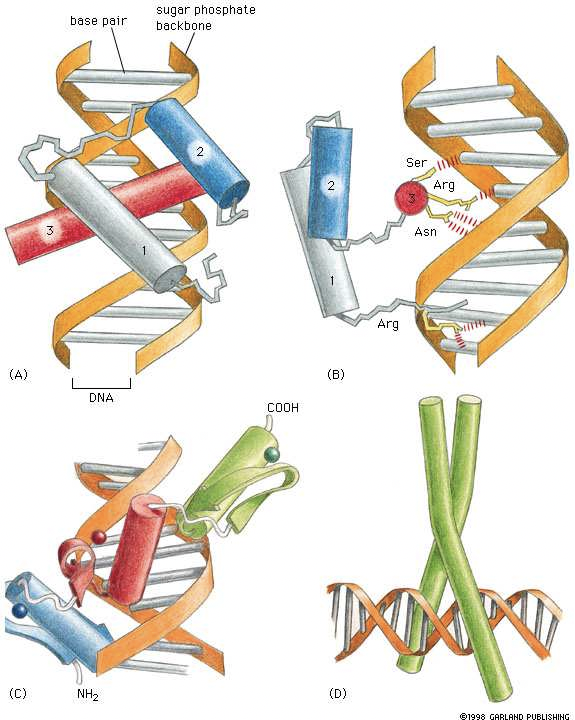
Antes de poder adentrarnos en algoritmos para el descubrimiento de motivos, primero debemos entender las características de los motivos, especialmente aquellos que hacen que los motivos sean algo difíciles de encontrar. Como se mencionó anteriormente, los motivos son generalmente muy cortos, generalmente de solo 6-8 pares de bases de largo. Adicionalmente, los motivos pueden ser degenerados, donde solo los nucleótidos en ciertas ubicaciones dentro del motivo afectan la función del motivo. Esta degeneración surge porque los factores de transcripción son libres de interactuar con sus motivos correspondientes de maneras más complejas que una simple relación de complementariedad. Como se ve en 17.1, muchas proteínas interactúan con el motivo no abriendo el ADN para verificar la complementariedad de bases, sino explorando los espacios, o surcos, entre las dos cadenas principales de fosfato de azúcar. Dependiendo de la estructura física del factor de transcripción, la proteína solo puede ser sensible a la diferencia entre purinas y pirimidinas o bases débiles y fuertes, a diferencia de identificar pares de bases específicos. La topología del factor de transcripción puede incluso hacerla tal que ciertos nucleótidos no interactúen en absoluto, permitiendo que esas bases actúen como comodines.
Este tema de degeneración dentro de un motivo plantea un problema desafiante. Si solo estuviéramos buscando un k-mer fijo, simplemente podríamos buscar el k-mer en todas las secuencias que estamos viendo usando alineación local
© Garland Publishing. Todos los derechos reservados. Este contenido está excluido de nuestra licencia Creative Commons. Para obtener más información, consulte http://ocw.mit.edu/help/faq-fair-use/.
herramientas. Sin embargo, el motivo puede variar de una secuencia a otra. Debido a esto, se dice que una cadena de nucleótidos que se sabe que es un motivo regulador es una instancia de un motivo porque representa una de las posibles muchas combinaciones diferentes de nucleótidos que cumplen la función del motivo.
En nuestros enfoques, hacemos dos suposiciones sobre los datos. Primero, suponemos que no hay correlaciones por pares entre bases, es decir, que cada base es independiente de cada otra base. Si bien tales correlaciones existen en la vida real, considerarlas en nuestro análisis conduciría a un crecimiento exponencial del espacio de parámetros que se está considerando, y consecuentemente correríamos el riesgo de sobreajustar nuestros datos. La segunda suposición que hacemos es que todos los motivos tienen longitudes fijas; de hecho, esta aproximación simplifica enormemente el problema. Sin embargo, incluso con estos dos supuestos, el hallazgo de motivos sigue siendo un problema muy desafiante. El tamaño relativamente pequeño de los motivos, junto con su gran variedad, hace bastante difícil localizarlos. Además, la ubicación de un motivo en relación con el gen correspondiente está lejos de ser fija; el motivo puede estar aguas arriba o aguas abajo, y la distancia entre el gen y el motivo también varía. De hecho, a veces el motivo está lejos de 10k a 10M pares de bases del gen.
Los motivos resumen la especificidad de la secuencia de TF
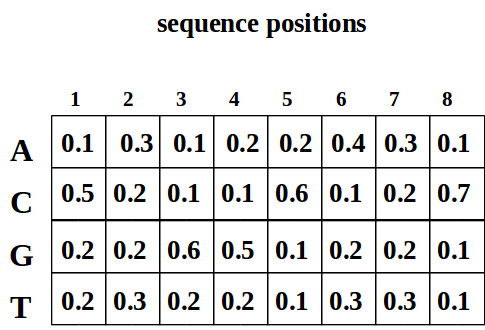
Debido a que las instancias de motivos exhiben gran variedad, generalmente usamos una Matriz de Peso de Posición (PWM) para caracterizar el motivo. Esta matriz da la frecuencia de cada base en cada ubicación del motivo. La siguiente figura muestra un ejemplo de PWM, donde pck corresponde a la frecuencia de la base c en la posición k dentro del motivo, con p c0 denotando la distribución de bases en regiones no motivadas.
Ahora definimos el problema del hallazgo de motivos de manera más rigurosa. Suponemos que se nos da un conjunto de genes corregulados y funcionalmente relacionados. Muchos motivos fueron descubiertos previamente haciendo huella
experimentos, que aíslan secuencias unidas por factores de transcripción específicos, y por lo tanto es más probable que correspondan a motivos. Existen varios métodos computacionales que se pueden utilizar para localizar motivos:
- Realice una alineación local a través del conjunto de secuencias y explore las alineaciones que resultaron en una puntuación de alineación muy alta.
- Modele las regiones promotoras usando un modelo oculto de Markov y luego use un modelo generativo para encontrar secuencias no aleatorias.
- Reducir el espacio de búsqueda aplicando conocimientos previos sobre cómo deberían verse los motivos.
- Búsqueda de bloques conservados entre diferentes secuencias.
- Examine la frecuencia de kmers a través de regiones altamente propensas a contener un motivo.
- Utilice métodos probabilísticos, como EM, Gibbs Sampling o un algoritmo codicioso
El método 5, utilizando frecuencias relativas de kmer para descubrir motivos, presenta algunos desafíos a considerar. Por ejemplo, podría haber muchas palabras comunes que ocurren en estas regiones que de hecho no son motivos regulatorios sino diferentes conjuntos de instrucciones. Además, dada una lista de palabras que podrían ser un motivo, no es seguro que el motivo más probable sea la palabra más común; por ejemplo, mientras que los motivos generalmente están sobrerrepresentados en las regiones promotoras, los factores de transcripción pueden ser incapaces de unirse si hay un exceso de motivos presentes. Una posible solución a este problema podría ser encontrar kmers con frecuencia relativa máxima en las regiones promotoras en comparación con las regiones de fondo. Esta estrategia se realiza comúnmente como una etapa de post-procesamiento para reducir el número de motivos posibles.
En la siguiente sección, hablaremos más sobre estos algoritmos probabilísticos así como los métodos para usar la frecuencia kmer para el descubrimiento de motivos. También volveremos a la idea de usar kmers para encontrar motivos en el contexto del uso de la conservación evolutiva para el descubrimiento de motivos.