29.5: Evolución Humana
- Page ID
- 53923
No es sorprendente que la comunidad científica tenga una larga y algo polémica historia de interés en la dinámica poblacional reciente. Si bien efectivamente parte de este interés se aplicó hacia fines más nefastos, como las justificaciones científicas del racismo para la eugenesia pero éstas son cada vez más la excepción y no la regla. Los primeros estudios de la dinámica poblacional fueron primitivos en muchos aspectos. La cuantificación de las diferencias entre las poblaciones humanas se realizó originalmente utilizando tipos de sangre, ya que parecían ser fenotípicamente neutros, podrían probarse para fuera del cuerpo y parecían ser polimórficos en muchas poblaciones humanas diferentes. Avance rápido hasta el presente, y la comunidad científica se ha dado cuenta de que existen otras glicoproteínas más allá de los grupos sanguíneos A, B y O que son mucho más polimórficas en la población. A medida que la ciencia continuó avanzando y la secuenciación se hizo realidad, comenzaron la secuenciación del genoma completo del cromosoma Y, marcadores mitocondriales y microsatélites alrededor de ellos. ¿Qué tienen de especial esos dos tipos de datos genéticos? En primer lugar, son bastante cortos por lo que se pueden secuenciar más fácilmente que otros cromosomas. Más allá del tamaño, la razón por la que los cromosomas Y y mitocondriales fueron de tal interés es porque no se recombinan, y pueden ser utilizados para reconstruir fácilmente árboles heredados. Esto es precisamente lo que hace que estos cromosomas sean especiales en relación con un trozo corto en un autosoma; sabemos exactamente de dónde viene porque podemos rastrear el linaje paterno o materno hacia atrás en el tiempo.
Este tipo de reconstrucción no funciona con otros cromosomas. Si uno fuera a generar un árbol usando cierto trozo de todo el cromosoma 1 en una determinada población, por ejemplo, de hecho formarían una filogenia pero esa filogenia sería escogida de ancestros aleatorios en cada uno de los árboles genealógicos.
A medida que la secuenciación continuaba desarrollándose y haciéndose más efectiva, se estaba proponiendo el proyecto del genoma humano, y junto con él hubo un fuerte impulso para incluir algún tipo de medida de diversidad en los datos genómicos. Técnicamente hablando, fue más fácil simplemente mirar los microsatélites para esta medida de diversidad porque se pueden estudiar en gel para ver polimorfismos de tamaño en lugar de inspeccionar un polimorfismo de secuencia. Como recordatorio, un microsatélite es una región de longitud variable en el genoma humano a menudo caracterizada por repeticiones cortas en tándem. Una razón para los microsatélites son los retrovirus que se insertan en el genoma, como los elementos ALU en el genoma humano. Estos elementos a veces se vuelven activos y se retrotransponen como eventos de inserción y uno puede rastrear cuándo esos eventos de inserción han ocurrido en el linaje humano. De ahí que hubo un impulso, desde el principio, para ensayar estas partes del genoma en una variedad de poblaciones diferentes. Lo realmente atractivo de los microsatélites es que son altamente polimórficos y en realidad se puede inferir su tasa de mutación. De ahí que no sólo podamos decir que existe una cierta relación entre las poblaciones en base a estas tasas, sino que también podemos decir cuánto tiempo han estado evolucionando e incluso cuando ocurrieron ciertas mutaciones, y cuánto tiempo ha estado en ciertas ramas del árbol filogenético.
FAQ
P: ¿No se puede hacer esto simplemente con SNP?
R: No se puede hacer muy fácilmente con los SNP.
Puedes hacerte una idea de la edad que tienen en función de su frecuencia alélica, pero también van a ser influenciados por la selección.
Después del proyecto del genoma humano, vino el proyecto Hapmap de herencia de haplotipos que analizó el genoma de los SNP en todo el genoma. Hemos discutido en detalle la herencia de haplotipos en capítulos anteriores donde aprendimos la importancia de Hapmap en el diseño de matrices de genotipado que analizan SNP que marcan haplotipos comunes en la población.
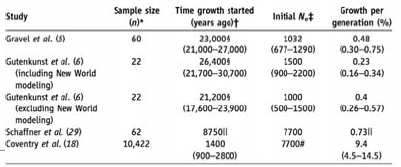
Los efectos de los cuellos de botella en la diversidad humana El uso de esta riqueza de datos a través de estudios y una plétora de técnicas matemáticas ha llevado a la constatación de que los humanos, de hecho, tienen una diversidad muy baja dada nuestra población censal; lo que implica un pequeño tamaño poblacional efectivo. Utilizando el modelo Wright-Fisher es posible trabajar desde el nivel de diversidad y el número de mutaciones que vemos en la población actual para generar un tamaño de población fundacional. Cuando se realiza este cómputo, resulta ser alrededor de 10,000.
FAQ
P: ¿Por qué es esto mucho más pequeño que el tamaño de nuestra población censal?
R: Había un cuello de botella poblacional en alguna parte.
La mayor parte de la variación total entre humanos ocurre dentro del continente. Se puede medir cuánta diversidad se explica por la geografía y cuánto no lo es. Resulta que la mayor parte no se explica por la geografía. De hecho, las variantes más comunes son polimórficas en cada población y si una variante común es única para una población determinada, probablemente no haya habido tiempo suficiente para que eso suceda por deriva misma. Recordemos lo poco probable que es llegar a una alta frecuencia alélica en el transcurso de varias generaciones solo por mera casualidad. De ahí que podamos interpretar esto como una señal de selección cuando ocurre. Toda la evidencia en términos de comparación de patrones de diversidad y árboles con haplotipos ancestrales converge en una hipótesis fuera de África que es el consenso abrumador en el campo y es la lente a través de la cual revisamos todos los datos genéticos de la población. Partiendo de la población fundadora africana, se han realizado trabajos que han demostrado que es posible modelar el crecimiento poblacional utilizando el modelo wright fisher. Los estudios han demostrado que la tasa de crecimiento que vemos en las poblaciones asiáticas y europeas solo es consistente con un gran crecimiento exponencial después del evento fuera de África.
Esto nos ayuda a comprender las razones de las diferencias fonotípicas entre las razas, ya que los cuellos de botella seguidos de un crecimiento exponencial pueden conducir a un exceso de alelos raros. La presente teoría sobre la diversidad humana establece que hubo eventos secundarios de cuello de botella después de que la población fundadora emigró fuera de África. Estos fundadores fueron, en algún momento anterior, sujetos a un evento de cuello de botella aún menor que ahora se refleja en cada genoma humano del planeta, independientemente de su ascendencia inmediata. Es posible estimar qué tan pequeño era el cuello de botella original observando las diferencias entre individuos de origen africano y europeo, inferyendo los efectos del cuello de botella secundario, y el término de crecimiento exponencial de la población europea. La otra forma de acercarse a la estimación de eventos de cuello de botella es simplemente inspeccionar el espectro de frecuencias alélicas necesario para construir árboles coalescentes. De esta manera, uno puede tomar haplotipos a través del genoma y preguntar cuál fue el ancestro común más reciente observando cómo varía la coalescencia a lo largo del genoma. Por ejemplo, se puede adivinar que algún haplotipo fue seleccionado positivamente para solo recientemente dada la longitud del haplotipo. Un ejemplo de una de esas mutaciones recientes en la población europea es el gen de la lactasa. Otro ejemplo para la población asiática es el locus ER.
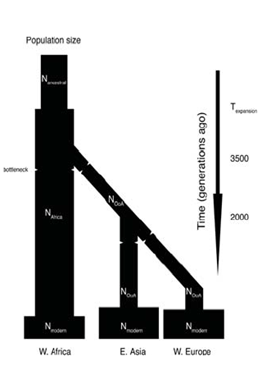
Hay una gran cantidad de literatura que muestra que cuando uno dibuja un árbol de coalescencia para la mayoría de los haplotipos termina yendo mucho antes cuando pensamos que ocurrió la especiación. Esto indica que ciertas características se han mantenido polimórficas durante mucho tiempo. Sin embargo, se puede observar esta distribución de características en todo el genoma e inferir algo sobre la historia de la población a partir de ella. Si hubo un cuello de botella reciente en una población, se verá reflejado por los antepasados siendo muy recientes mientras que cosas más antiguas habrán sobrevivido al cuello de botella. Se puede tomar la distribución de los tiempos de coalescencia y ejecutar simulaciones de cómo el efecto del tamaño de la población habría variado con el tiempo. El modelo para realizar este tipo de estudios fue delineado por Li y Durbin. La Figura 29.11 de su estudio ilustra dos eventos de cuello de botella de este tipo. El primero es el cuello de botella que se produjo en África mucho antes de las migraciones fuera del continente. A esto le siguió un cuello de botella específico de la población que resultó de grupos migratorios fuera de África. Esto se refleja en la diversidad de las poblaciones actuales en base a su ascendencia y se puede derivar de observar un par de cromosomas de dos personas cualesquiera en estas poblaciones.
Comprensión de la enfermedad
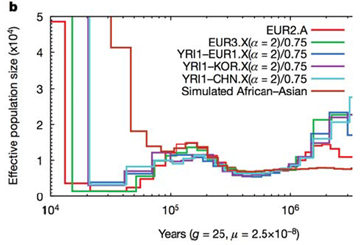
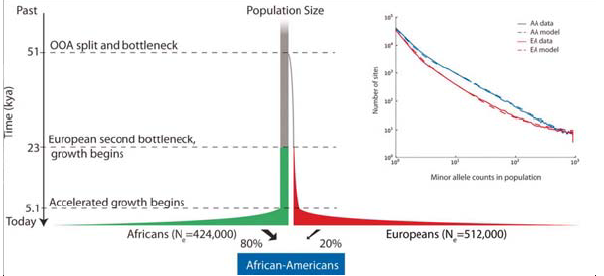
Comprender que las poblaciones humanas pasaron por cuellos de botella tiene implicaciones importantes para la enfermedad específica de la población subpermanente. Un estudio publicado por Tennessen et al. este año analizaba secuencias de exomas en muchas clases de individuos. El estudio tuvo como objetivo analizar cómo las variantes raras podrían estar contribuyendo a la enfermedad y, como consecuencia, fueron capaces de ajustar los modelos genéticos de poblaciones a los datos, y preguntar qué tipo de variantes deletéreas se vieron al secuenciar exomas de un amplio panel poblacional. Con este enfoque, fueron capaces de generar parámetros que describen cuánto tiempo hace que se produjo el crecimiento exponencial entre el fundador y las poblaciones ramificadas. Vea la figura 29.12 a continuación para una ilustración de esto:
Comprensión de la mezcla poblacional reciente
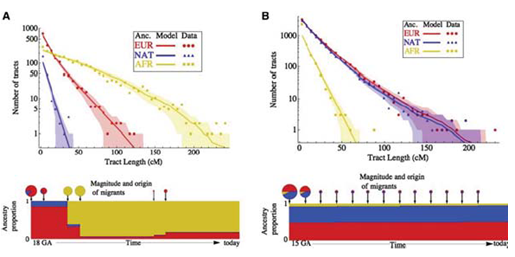
Además de ver los tiempos de coalescencia, también se puede realizar Análisis de Componentes Principales en SNP para comprender las mezclas poblacionales más recientes. Ejecutar esto en la mayoría de las poblaciones muestra agrupamiento con respecto a la ubicación geográfica. Hay algunas poblaciones, sin embargo, que experimentaron una mezcla reciente por razones históricas. Los dos más comúnmente referidos en la literatura científica son: los afroamericanos, que en promedio son 20
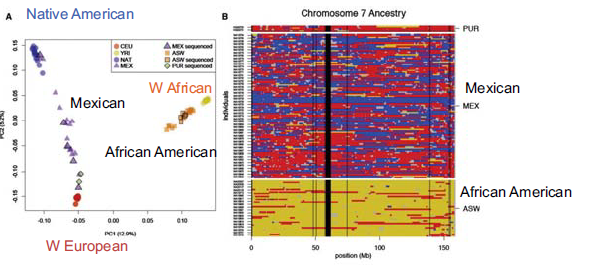
Hay dos cosas importantes que uno puede decir sobre el evento de mezcla de afroamericanos y mexicoamericanos. El primero y más obvio es inferir el nivel de mezcla. El segundo, y más interesante, es inferir cuándo ocurrió el evento de mezcla basado en el nivel real de mezcla. Como hemos comentado en capítulos anteriores, los significantes raciales del genoma se descomponen con la mezcla a causa de la recombinación en cada generación. Si la población está contenida, el porcentaje de aquellos con origen europeo y de África Occidental debería permanecer igual en cada generación, pero los segmentos se acortarán, debido a la mezcla. De ahí que la longitud de los bloques de haplotipos se pueda utilizar para remontarse a cuando se produjo originalmente la mezcla. (Cuando sucedió originalmente esperaríamos trozos grandes, siendo algunos gambits enteramente de origen africano, por ejemplo). Con este enfoque, se puede observar la distribución de las trampas de ascendencia recientes y luego ajustar un modelo al momento en que estos migrantes ingresaron a una población ancestral como se muestra a continuación: