
1: Laboratorio de Ortología/Paralog
- Page ID
- 52816

Para esta tarea girarás en tu alineación, tu árbol, los nombres de un par de ortólogos, los nombres de un par de parálogos, y una breve descripción del significado del gen que elegiste. Adicionalmente, tendrás un “Apéndice” con notas sobre los pasos (si sucedió algo en un paso que no fue descrito en las instrucciones, si notaste algo interesante).
INSTRUCCIONES
Obtener su conjunto de secuencias
- Consigue tus secuencias de “cebo”.
- Ve a Uniprot y escribe el nombre YFG + una especie modelo (por ejemplo, si estás interesado en genes de ciempiés en última instancia, pondrías en Drosophila). Lee un poco sobre tu gen y desplázate hacia abajo hasta FASTA. Da click en esto y te llevará a la secuencia proteica formateada FASTA.
- Si hay más de una “versión” de YFG para esa especie, obtenga la FASTA para todas ellas; probablemente sean homólogos. Por ejemplo, mirando a Dlx (el Distalless en vertebrados) encuentro 5 versión en humanos. Entonces obtendría la FASTA por cada una de estas. Ponlos todos en el mismo documento de texto.
- Ve a Uniprot y escribe el nombre YFG + una especie modelo (por ejemplo, si estás interesado en genes de ciempiés en última instancia, pondrías en Drosophila). Lee un poco sobre tu gen y desplázate hacia abajo hasta FASTA. Da click en esto y te llevará a la secuencia proteica formateada FASTA.
- Obtenga sus secuencias “modelo”. Sigue las mismas instrucciones para tus otros dos (o tres) modelos. Si tienes problemas para seleccionar tus modelos, ¡puedes pedirme ayuda! Agréguelos al documento de texto FASTA.
- Obtenga sus secuencias de “prueba”
- Explosión de su secuencia de “cebo” en NCBI blastp. Limita tu búsqueda a tu especie de prueba, ayuda si conoces el nombre científico (Wikipedia tiene estos). Elija las secuencias con un valor inferior a 1e-10. Si hay demasiados, solo elige el top 5. Si no hay ninguno con un evalue tan bajo, elige el top 3. Agréguelos al documento de texto FASTA.
- Obtenga su secuencia “outgroup”
- Esta debe ser una secuencia que sea similar pero no homóloga a YFG. Para encontrar uno, vuelve a UniProt y da clic en BLAST. Pegue en su secuencia de cebo y elija la base de datos UniRef50 en el menú desplegable y haga clic en ejecutar. Espera.
- Desplácese hacia abajo en la lista de resultados hasta que comience a ver nombres de genes que difieren de YFG. Haga clic en uno de estos de alta puntuación y obtenga la secuencia FASTA, agregue esto a su documento de texto FASTA en la parte superior.
Haciendo tu árbol
El siguiente paso es alinear tus secuencias y hacer un árbol. La alineación de secuencias coloca las partes más similares de cada secuencia en columnas verticales. Hace más fácil ver visualmente si las secuencias son realmente muy similares o no tanto. También a veces se pueden ver cosas como dominios conservados en una alineación. La otra cosa útil de las alineaciones es que pueden ser utilizadas para puntuar similitud mediante algoritmos estadísticos. Estos programas utilizan alineaciones para inferir relaciones filogenéticas.
- Pegue su secuencia formateada FASTA en https://www.ebi.ac.uk/Tools/msa/muscle/ y elija Pearson/Fasta como salida. La salida es importante porque necesitamos un archivo de salida que el siguiente programa pueda leer.
- La página de resultados tiene un montón de opciones diferentes. Guarde la versión básica de texto y luego haga clic para visualizar su alineación de diferentes maneras. ¿Ves algo interesante? ¿Algún patrón?
- Sube este “archivo de alineación” a http://iqtree.cibiv.univie.ac.at. y haz clic en enviar trabajo. IQtree por defecto probará diferentes modelos de evolución molecular en sus datos y verá cuál encaja. No vamos a usar modelos súper elegantes, así que no necesitamos agregar cosas como una distribución gamma o heterogeneidad de tasa libre. A medida que IQTree corre puedes reflexionar sobre la diferencia entre parálogos y ortólogos y/o comenzar a escribir una descripción de tu gen y lo que sucedió en cada paso que hiciste para enterarte de su complejidad genética en tu organismo no modelo.
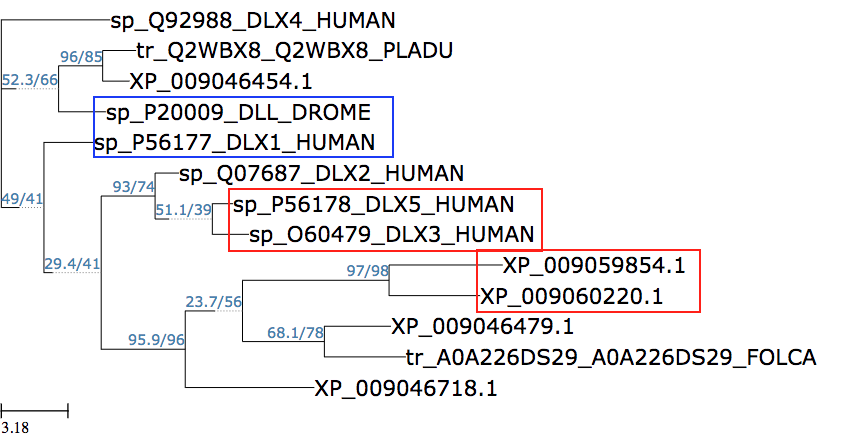 |
Salida de muestra de Hyung Joo Kim y Kinsei Imada Este árbol busca averiguar si el gen de la familia Distalless de Drosophila INDY (I'm Not Dead Yet) tiene un homólogo en un insecto relacionado, Folsomia candida (FOLCA). Los parálogos humanos están enmarcados en rojo. El pariente más cercano en este árbol a Drosophila INDY (XP_009059854.1) es un gen de moluscos (ortólogos enmarcados en rojo). El gen FOLCA se encuentra dentro del mismo clado que el INDY de Drosophila, sugiriendo que podría ser un homólogo de INDY. En azul están humanos y Drosophila representantes de un evento de duplicación de genes ancestrales. |