
1.3: Introducción - Agua y Buffers
- Page ID
- 53315

Fuente: BiochemFFA_1_3.pdf. Todo el libro de texto está disponible de forma gratuita de los autores en http://biochem.science.oregonstate.edu/content/biochemistry-free-and-easy

Cuando se trata de agua, literalmente nos estamos ahogando en ella, ya que el agua es, con mucho, el componente más abundante de cada célula. Para entender la vida, comenzamos la discusión con los fundamentos del agua, porque todo lo que sucede en las células, incluso las reacciones enterradas en lo profundo de las enzimas, lejos del agua, está influenciado por la química del agua.
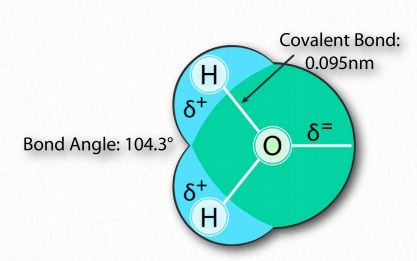
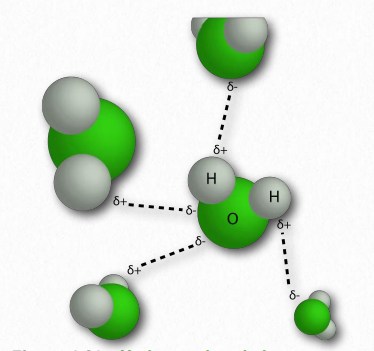
La molécula de agua tiene forma amplia de 'V' (el ángulo HO-H es 104°) con distribución desigual de electrones entre los átomos de oxígeno e hidrógeno (Figura 1.23). El oxígeno, con su mayor electronegatividad, mantiene los electrones más cerca de sí mismo que los hidrógenos. Los hidrógenos, como resultado, se describen como que tienen una carga positiva parcial (típicamente designada como δ+) y el oxígeno tiene una carga negativa parcial (escrita como δ-). Así, el agua es una molécula polar porque las cargas se distribuyen a su alrededor de manera desigual, no simétrica.
El agua como disolvente
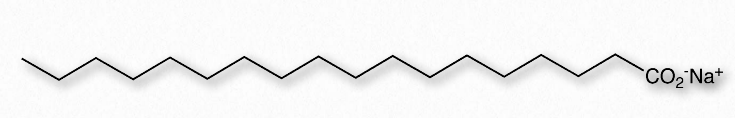
El agua (Figura 1.23) se describe como un solvente debido a su capacidad para solvatar (disolver) muchas, pero no todas, moléculas. Las moléculas que son iónicas o polares se disuelven fácilmente en el agua, pero las sustancias no polares se disuelven mal en el agua, si acaso. El petróleo, por ejemplo, que es no polar, se separa del agua cuando se mezcla con él. Por otro lado, el cloruro de sodio, que ioniza, y el etanol, que es polar, son capaces de formar enlaces de hidrógeno, por lo que ambos se disuelven en agua. La solubilidad del etanol en agua es crucial para cerveceros, enólogos y destiladores, pero para esta propiedad, no habría vino, cerveza ni licores. Como se explicó en una sección anterior, utilizamos el término hidrófilo para describir sustancias que interactúan bien con el agua y se disuelven en ella y el término hidrófobo para referirse a materiales que son no polares y no se disuelven en el agua. En el cuadro 1.3 se muestran algunas sustancias polares y no polares. Un tercer término, anfifílico, se refiere a compuestos que tienen ambas propiedades. Los jabones, por ejemplo, son anfifílicos, que contienen una cola alifática larga, no polar y una cabeza que ioniza.

Cuadro 1.3 Imagen de Aleia Kim
Solubilidad
La solubilidad de los materiales en agua se basa en cambios de energía libre, medidos por ΔG. Recuerde, de la química, que H es la entalpía (calor a presión constante) y S es la entropía. Ante esto,
\[ΔG = ΔH - TΔS\]
donde T es la temperatura en Kelvin. Para que un proceso sea favorable, el ΔG para ello debe ser menor que cero.
A partir de la ecuación, se favorecerán los valores reducidos de ΔG con disminuciones en la entalpía y/o aumentos en la entropía. Consideremos primero por qué los materiales no polares no se disuelven en el agua. Podríamos imaginar una situación en la que el proceso de disolución involucra el “rodeamiento” de cada molécula del soluto no polar en el agua, al igual que cada sodio y cada ion cloruro se rodea de moléculas de agua a medida que la sal se disuelve.
Organización del agua
Hay una diferencia significativa, aunque entre rodear una molécula no polar con moléculas de agua y los iones circundantes (o compuestos polares) con moléculas de agua.
La diferencia es que dado que las moléculas no polares realmente no interactúan con el agua, el agua se comporta de manera muy diferente que con iones o moléculas que forman enlaces de hidrógeno. De hecho, alrededor de cada molécula no polar, el agua se organiza muy, alineándose regularmente. Como cualquier estudiante de primer año de química probablemente recuerda, la entropía es una medida del trastorno, por lo que cuando algo se ordena, la entropía disminuye, es decir, el ΔS es negativo, por lo que el término TΔS en la ecuación es positivo (negativo de un negativo).
Dado que la mezcla de una sustancia no polar con agua generalmente no tiene ningún componente térmico significativo, el ΔG es positivo. Esto significa, entonces, que disolver un compuesto no polar en agua no es favorable y no ocurre en ningún grado significativo. Además, cuando el material no polar se asocia consigo mismo y no con el agua, entonces las moléculas de agua son libres de mezclarse, sin ser ordenadas, resultando en un aumento de la entropía. Por lo tanto, la entropía impulsa la separación de sustancias no polares de las soluciones acuosas.

Sustancias anfifílicas
A continuación, consideramos mezclar una sustancia anfifílica, como un jabón, con agua (Figura 1.24). Los iones de sodio unidos a los ácidos grasos en el jabón se desprenden fácilmente en solución acuosa, dejando atrás una molécula cargada negativamente en un extremo y una región no polar en el otro extremo. La ionización del jabón provoca un aumento de la entropía, dos partículas en lugar de una. La porción no polar del ion jabón cargado negativamente es problemática; si se expone al agua, hará que el agua se organice y dará como resultado una disminución de la entropía y un ΔG positivo.
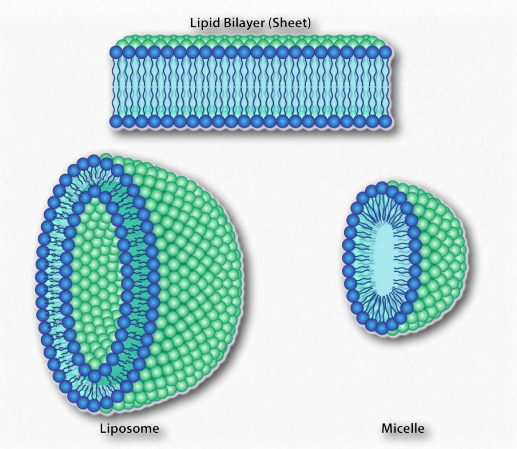
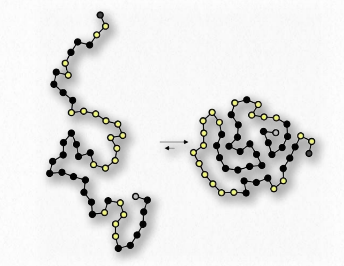
Como sabemos que los ácidos grasos se disuelven en el agua, debe haber algo más en juego. La hay. Al igual que las moléculas no polares en el primer ejemplo asociadas entre sí y no con agua, también las porciones no polares de los iones de jabón se asocian entre sí y excluyen el agua. El resultado es que los iones de jabón se disponen como micelas (Figura 1.25) con las porciones no polares en el interior de la estructura alejadas del agua y las porciones polares en el exterior interactuando con el agua.
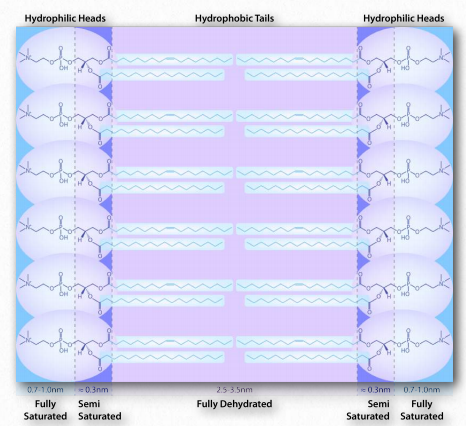
La interacción de las cabezas polares con el agua devuelve el agua a su estado más desordenado. Este incremento en el trastorno, o entropía, impulsa la formación de micelas. Como se verá en la discusión de la bicapa lipídica, las mismas fuerzas impulsan a los glicerofosfólidos y esfingolípidos a formar espontáneamente bicapas donde las porciones no polares de las moléculas interactúan entre sí para excluir el agua y las porciones polares se disponen en el exterior de la bicapa ( Figura 1.28).
Otro ejemplo más se ve en el plegamiento de proteínas globulares en el citoplasma. Los aminoácidos no polares se encuentran en la porción interior de la proteína (excluida el agua). La interacción de los aminoácidos no polares resulta ser una fuerza impulsora para el plegamiento de las proteínas ya que se están haciendo en una solución acuosa.
Enlaces de hidrógeno
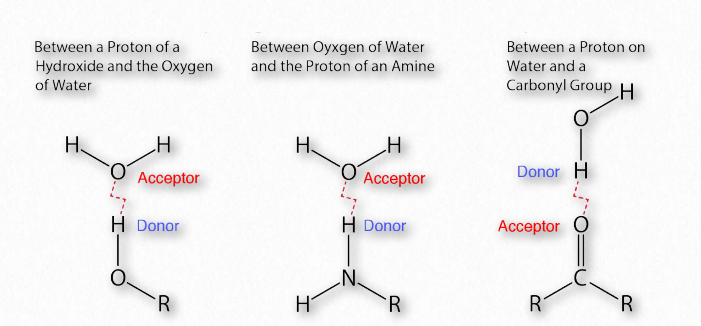
La importancia de los enlaces de hidrógeno en la bioquímica (Figura 1.30) es difícil de exagerar. El propio Linus Pauling dijo:
“..... Creo que a medida que los métodos de la química estructural se aplican más a problemas fisiológicos se encontrará que la significación del enlace de hidrógeno para la fisiología es mayor que la de cualquier otra característica estructural única”.
En 2011, un grupo de tareas de la IUPAC dio una definición basada en pruebas de los enlaces de hidrógeno que establece,
“El enlace de hidrógeno es una interacción atractiva entre un átomo de hidrógeno de una molécula o un fragmento molecular X—H en el que X es más electronegativo que H, y un átomo o un grupo de átomos en la misma o en una molécula diferente, en el que hay evidencia de formación de enlaces”.
Cargos Parciales
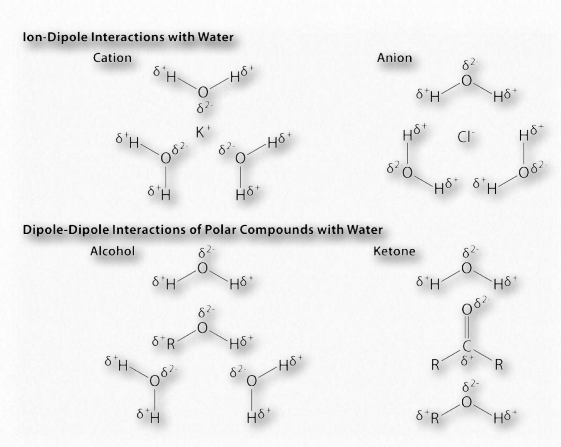
La diferencia de electronegatividad entre el hidrógeno y la molécula a la que está unido covalentemente da lugar a cargas parciales como se describió anteriormente. Estas pequeñas cargas (δ+ y δ-) dan como resultado la formación de enlaces de hidrógeno, los cuales ocurren cuando la carga positiva parcial de un átomo de hidrógeno es atraída a la negativa parcial de otra molécula. En el agua, eso significa que el hidrógeno de una molécula de agua es atraído por el oxígeno de otra (Figura 1.31). Dado que el agua es una molécula asimétrica, significa también que las cargas son asimétricas. Una distribución tan desigual es lo que hace que un dipolo. Las moléculas dipolares son importantes para las interacciones con otras moléculas dipolares y para disolver sustancias iónicas (Figura 1.32).
Los enlaces de hidrógeno no son exclusivos del agua. De hecho, son fuerzas importantes que mantienen unidas macromoléculas que incluyen proteínas y ácidos nucleicos. Los enlaces de hidrógeno ocurren dentro y entre macromoléculas.
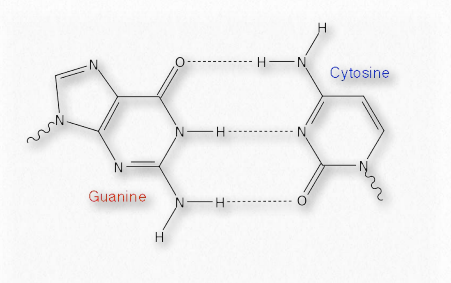
El emparejamiento complementario que ocurre entre bases en cadenas opuestas de ADN, por ejemplo, se basa en enlaces de hidrógeno. Cada enlace de hidrógeno es relativamente débil (comparado con un enlace covalente, por ejemplo - Cuadro 1.4), pero colectivamente pueden ser bastante fuertes.

Cuadro 1.4 Imagen de Aleia Kim
Beneficios de interacciones débiles
Su debilidad, sin embargo, en realidad es bastante beneficiosa para las células, particularmente en lo que respecta a los ácidos nucleicos (Figura 1.33). Las cadenas de ADN, por ejemplo, deben separarse en tramos cortos en los procesos de replicación y síntesis de ARN. Dado que solo unos pocos pares de bases a la vez necesitan ser separados, la energía requerida para hacer esto es pequeña y las enzimas involucradas en los procesos pueden separarlos fácilmente, según sea necesario. Los enlaces de hidrógeno también juegan un papel en la unión de sustratos a enzimas, catálisis e interacción proteína-proteína, así como otros tipos de unión, como proteína-ADN o anticuerpo-antígeno.
Como se señaló, los enlaces de hidrógeno son más débiles que los enlaces covalentes (Cuadro 1.4) y su fuerza varía de muy débil (1-2 kJ/mol) a bastante fuertes (29 kJ/mol). Los enlaces de hidrógeno solo ocurren en distancias relativamente cortas (2.2 a 4.0 Å). Cuanto más alejada esté la distancia del enlace de hidrógeno, más débil es el enlace.
La fuerza del enlace en kJ/mol representa la cantidad de calor que se debe poner en el sistema para romper el enlace; cuanto mayor sea el número, mayor será la fuerza del enlace. Los enlaces de hidrógeno se rompen fácilmente usando calor. La ebullición del agua, por ejemplo, requiere la ruptura de los enlaces H. Cuando una estructura biológica, como una proteína o una molécula de ADN, se estabiliza por enlaces de hidrógeno, romper esos enlaces desestabiliza la estructura y puede resultar en la desnaturalización de la sustancia, pérdida de estructura. Es en parte por esta razón que la mayoría de las proteínas y todos los ADN pierden sus estructuras nativas, o plegadas, cuando se calientan a ebullición.
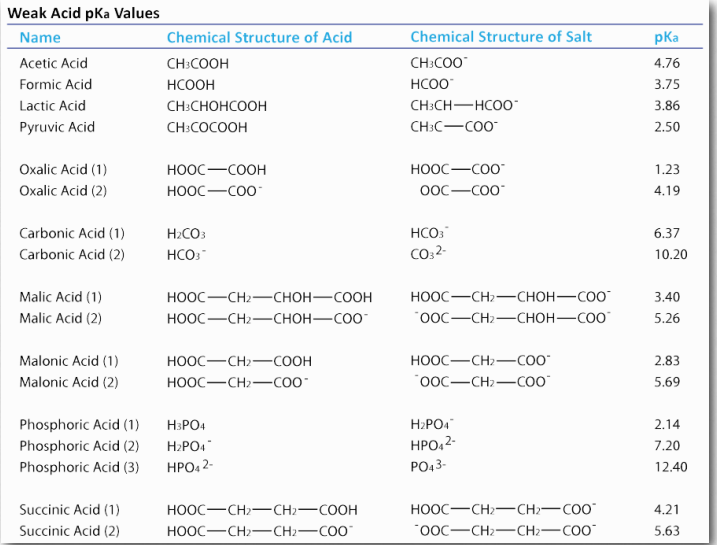
Imagen de Aleia Kim Table 1.5
Para las moléculas de ADN, la desnaturalización da como resultado la separación completa de las cadenas entre sí. Para la mayoría de las proteínas, esto significa pérdida de su característica estructura tridimensional y con ella, pérdida de la función que desempeñaron. Aunque algunas proteínas pueden tranquilizar fácilmente su estructura original cuando se enfría la solución en la que se encuentran, la mayoría no puede, esta es una de las razones por las que cocinamos nuestros alimentos. Las proteínas son esenciales para la vida, por lo que la desnaturalización de las proteínas bacterianas resulta en la muerte de cualquier microorganismo que contamine los alimentos.
La importancia de los búferes
El agua puede ionizarse en un grado leve (10-7 M) para formar H+ (protón) y OH- (hidróxido). Medimos la concentración de protones de una solución con pH, que es el log negativo de la concentración de protones.
pH = -Log [H+]
Si la concentración de protones, [H+] = 10-7 M, entonces el pH es 7. Podríamos medir fácilmente la concentración de hidróxido con el PoH mediante la ecuación paralela,
PoH = -Registro [OH -]
En agua pura, la disociación de un protón crea simultáneamente un hidróxido, por lo que el PoH del agua pura es 7, también. Esto también significa que
pH+ PoH = 14
Ahora bien, debido a que los protones e hidróxidos pueden combinarse para formar agua, una gran cantidad de uno hará que haya una pequeña cantidad del otro. ¿Por qué es este el caso? En términos simples, si vuelco 0.1 moles de H+ en una solución de agua pura, la alta concentración de protones reaccionará con la cantidad relativamente pequeña de hidróxidos para crear agua, reduciendo así la concentración de hidróxido. De igual manera, si vuelco el exceso de hidróxido (como NaOH, por ejemplo) en agua pura, la concentración de protones cae por la misma razón.
Ácidos vs bases
Los químicos utilizan el término “ácido” para referirse a una sustancia que tiene protones que pueden disociarse (desprenderse) cuando se disuelven en agua. Utilizan el término “base” para referirse a una sustancia que puede absorber protones cuando se disuelve en agua. Tanto los ácidos como las bases vienen en formas fuertes y débiles. (Los ejemplos de ácidos débiles se muestran en la Tabla 1.5.) Los ácidos fuertes, como el HCl, se disocian completamente en agua. Si agregamos 0.1 moles (6.02x1022 moléculas) de HCl a una solución para hacer un litro, tendrá 0.1 moles de H+ y 0.1 moles de Cl- o 6.02x1022 moléculas de cada una. No habrá HCl restante cuando esto suceda. Una base fuerte como el NaOH también se disocia completamente en Na+ y OH-.

Ácidos Débiles
Los ácidos y bases débiles difieren de sus contrapartes fuertes. Cuando pones un mol de ácido acético (HAc) en agua pura, solo un pequeño porcentaje de las moléculas de HAc se disocian en H+ y Ac-. Claramente, los ácidos débiles son muy diferentes de los ácidos fuertes. Las bases débiles se comportan de manera similar, salvo que aceptan protones, en lugar de donarlos. Ya que podemos ver todo como una forma de un ácido débil, aquí no vamos a utilizar el término base débil.

Los estudiantes suelen estar desconcertados y esperan que [H +] = [A -] porque la ecuación de disociación muestra uno de cada uno de HA. Esto es, de hecho, cierto SÓLO cuando se permite que el HA se disocie en agua pura. Por lo general, el HA se coloca en solución que tiene protones e hidróxidos para afectar las cosas. Esos protones y/o hidróxidos cambian el H+ y la Aconcentración de manera desigual, ya que A- puede absorber algunos de los protones y/o HA puede liberar H+ cuando está influenciado por el OH- en la solución. Por lo tanto, se debe calcular la concentración de protones a partir del pH usando la ecuación de Henderson Hasselbalch.
\[pH = pKa + log ([Ac- ]/[HAc])\]

Imagen de Aleia Kim Table 1.6
Quizás te preguntes por qué nos importan los ácidos débiles. Puede que nunca hayas pensado mucho en los ácidos débiles cuando estabas en Química General. Tu instructor los describió como tampones y probablemente memorizaste obedientemente el hecho de que “los tampones son sustancias que resisten el cambio de pH” sin realmente aprender qué significaba Clearing Confusión, esto significaba. Los búferes son demasiado importantes para ser pensados de esta manera.
UPS
Los ácidos débiles son críticos para la vida porque su afinidad por los protones hace que se comporten como un SAI. No nos referimos al UPS que es el United Parcel Service, sino a los sistemas de respaldo de batería encerrado para computadoras llamadas Fuentes de alimentación ininterrumpida que se ponen en marcha para mantener una computadora funcionando durante una falla eléctrica. La batería en una computadora portátil es un UPS, por ejemplo.
Podemos pensar en los ácidos débiles como Proveedores de Protones Ininterrumpibles dentro de ciertos rangos de pH, proporcionando (o absorbiendo) protones según sea necesario. Los ácidos débiles ayudan a mantener la concentración de H+ (y por lo tanto el pH) de la solución en la que están relativamente constantes.
Considera el sistema bicarbonato/ácido carbónico. La Figura 1.35 muestra lo que sucede cuando H 2 CO 3 se disocia. La adición de iones hidróxido (al agregar una base fuerte como NaOH) a la solución hace que los iones H+ reaccionen con los iones OH- para hacer agua. En consecuencia, la concentración de iones H+ bajaría y el pH subiría.
Sin embargo, en contraste con la situación con una solución de agua pura, existe una fuente de respaldo de H+ disponible en forma de H 2 CO 3. Aquí es donde entra en juego la función UPS. A medida que los protones son quitados por los iones hidroxilo añadidos (haciendo agua), son reemplazados en parte por protones del H 2 CO 3. Es por ello que un ácido débil es un tampón. Resiste los cambios en el pH liberando protones para compensar aquellos “agotados” en la reacción con los iones hidroxilo.

Henderson-Hasselbalch
Es útil para poder predecir la respuesta del sistema H 2 CO 3 a los cambios en la concentración de H+. La ecuación de Henderson-Hasselbalch define la relación entre el pH y la relación de HCO 3 - y H 2 CO 3. Es
pH = pKa + log ([HCO 3 -]/[H 2 CO 3])
Esta sencilla ecuación define la relación entre el pH de una solución y la relación de HCO 3 - y H 2 CO 3 en ella. El nuevo término, llamado pKa, se define como
pKa = -Log K a,
así como
pH = -Log [H +].
El Ka es la constante de disociación ácida y es una medida de la fuerza de un ácido. Para un ácido general, HA, que se disocia como
HA H + + A -, K a = [H +] [A -]/[HA]
Así, cuanto más fuerte sea el ácido, más protones se disociarán de él cuando se agreguen al agua y mayor será el valor que tendrá su Ka. Los valores grandes de Ka se traducen en valores más bajos de pKa. Como resultado, cuanto menor es el valor de pKa para un ácido dado, más fuerte es el ácido débil.
PkA constante
Tenga en cuenta que el pKa es una constante para un ácido dado. El pKa para el ácido carbónico es 6.37. En comparación, el pKa para el ácido fórmico es de 3.75. Por lo tanto, el ácido fórmico es un ácido más fuerte que el ácido acético. Un ácido más fuerte tendrá más protones disociados a un pH dado que un ácido más débil.
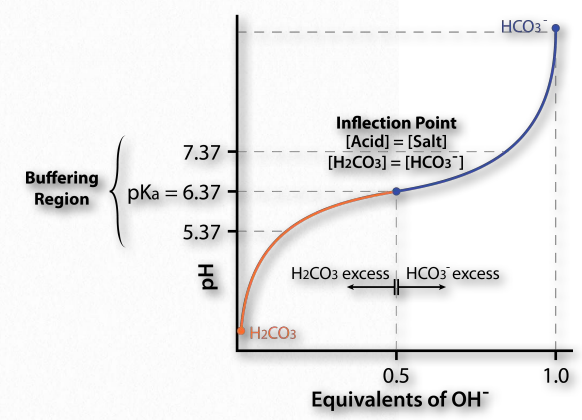
Ahora bien, ¿cómo se traduce esto en estabilizar el pH? La Figura 1.35 muestra una curva de titulación. En esta curva, la titulación comienza con las condiciones en la parte inferior izquierda (pH muy bajo). A este pH predomina la forma H 2 CO 3, pero a medida que se agrega más y más OH- (pasando a los 45 ¿Por qué nos importa el pH? Porque las moléculas biológicas pueden, en algunos casos, ser exquisitamente sensibles a los cambios en ella. A medida que cambia el pH de una solución, las cargas de las moléculas en la solución pueden cambiar, como verá. Cambiar las cargas en las moléculas biológicas, especialmente las proteínas, puede afectar drásticamente cómo funcionan e incluso si funcionan bien), el pH sube, sube la cantidad de HCO3- y (correspondientemente), baja la cantidad de H 2 CO 3. Observe que la curva “se aplana” cerca del pKa (6.37).
Región de almacenamiento en búfer
El aplanamiento de la curva nos dice es que el pH no está cambiando mucho (no subiendo tan rápido) como lo hizo antes cuando se añadió la misma cantidad de hidróxido. El sistema está resistiendo un cambio en el pH (no deteniendo el cambio, sino ralentizándolo) en la región de aproximadamente una unidad de pH por encima y una unidad de pH por debajo del pKa. Así, la región tampón del tampón ácido carbónico/ bicarbonato es de aproximadamente 5.37 a 7.37. Es máximamente fuerte a un pH de 6.37.
Ahora empieza a hacerse evidente cómo funciona el búfer. El HA puede donar protones cuando se necesitan extras (como cuando se agrega OH- a la solución mediante la adición de NaOH). De igual manera, A- puede aceptar protones cuando se agrega H+ extra a la solución (añadiendo HCl, por ejemplo). La máxima capacidad para donar o aceptar protones viene cuando
[A -] = [HA]
Esto es consistente con la ecuación de Henderson Hasselbalch y la curva de titulación. Cuando [A -] = [HA], pH = 6.37 + Log (1). Desde Log (1) = 0, pH = 6.37 = pKa para ácido carbónico. Así, para cualquier tampón, el tampón tendrá la máxima fuerza y exhibirá aplanamiento de su curva de titulación cuando [A -] = [HA] y cuando pH = pKa. Si un búfer tiene más de un pKa (Figura 1.36), entonces cada región pKa mostrará el comportamiento.
Buffered vs no buffer
Para entender qué tan bien protege un tampón contra los cambios en el pH, considere el efecto de agregar .01 moles de HCl a 1.0 litro de agua pura (sin cambio de volumen) a pH 7, en comparación con agregarlo a 1.0 litro de un tampón de acetato 1M a pH 4.76. Dado que el HCl se disocia completamente, en HCl 0.01M (10-2 M) tendrá 0.01M H+. Para el agua pura, el pH desciende de 7.0 a 2.0 (pH = -log (0.01M)).
Por el contrario, el pH del tampón acetato después de agregar la misma cantidad de HCl es de 4.74. Así, la solución de agua pura ve caer su pH de 7 a 2 (5 unidades de pH), mientras que la solución tamponada vio su caída de pH de 4.76 a 4.74 (0.02 unidades de pH). Claramente, el tampón minimiza el impacto de los protones agregados en comparación con el agua pura.
Capacidad de búfer
Es importante señalar que los tampones tienen capacidades limitadas por su concentración. Imaginemos que en el párrafo anterior, habíamos agregado los 0.01 moles de HCl a un tampón acetato que tenía una concentración de 0.01M y cantidades iguales de Ac- y HAc. Cuando tratamos de hacer las matemáticas en paralelo al cálculo anterior, vemos que hay 0.01M protones, pero sólo 0.005M A- para absorberlos. Podríamos imaginar que 0.005M de los protones serían absorbidos, pero eso aún dejaría 0.005M de protones sin amortiguar. Así, el pH de esta solución sería aproximadamente
pH = -log (0.005M) = 2.30
Al exceder la capacidad de tampón, el pH disminuyó significativamente en comparación con agregar la misma cantidad de protones a un tampón de acetato 1M. En consecuencia, al considerar los tampones, es importante reconocer que su concentración establece sus límites. Otro límite es el rango de pH en el que se espera controlar la concentración de protones.
Múltiples grupos ionizables
Ahora bien, ¿qué pasa si una molécula tiene dos (o más) grupos ionizables? Resulta, no en vano, que cada grupo tendrá su propio pKa y, como consecuencia, tendrá múltiples regiones de buffering.
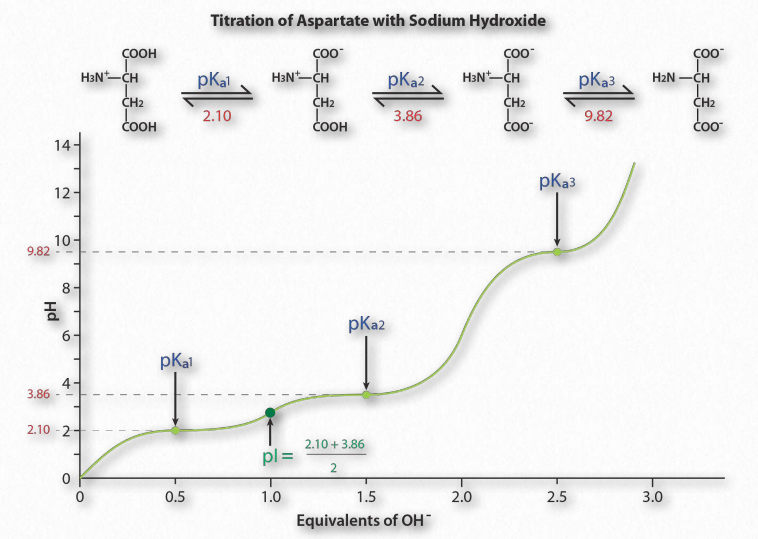
La Figura 1.36 muestra la curva de titulación para el aminoácido ácido aspártico. Tenga en cuenta que en lugar de un solo aplanamiento de la curva, como se observó para el ácido acético, la curva de titulación del ácido aspártico muestra tres regiones de este tipo. Estas son regiones tamponantes individuales, cada una centrada en los respectivos valores de pKa para el grupo carboxilo y el grupo amina.
El ácido aspártico tiene cuatro posibles cargas: +1 (grupo α-carboxilo, grupo α-amino y Rgrupo carboxilo tiene cada uno un protón), 0 (grupo α- carboxilo que falta protón, el grupo α- amino tiene un protón, el grupo R carboxilo tiene un protón), -1 (grupo α-carboxilo y grupo R carboxilo carecen cada uno de protón, el grupo α-amino retiene un protón), -2 (los grupos α-carboxilo, carboxilo del grupo R y α-amino carecen de protones adicionales).
Predicción
¿Cómo se predice la carga de un aminoácido a un pH dado? Una buena regla general para estimar la carga es que si el pH es más de una unidad por debajo del pKa para un grupo (carboxilo o amino), el protón está encendido. Si el pH es más de una unidad por encima del pKa para el grupo, el protón está apagado. Si el pH NO es más de una o menos de una unidad de pH del pKa, esta simple suposición no funcionará.
Además, es importante reconocer que estas reglas generales son únicamente estimaciones. El pI (pH al que la carga de una molécula es cero) es un valor exacto calculado como el promedio de los dos valores de pKa a cada lado de la región cero. Se calcula en el promedio de los dos valores de pKa alrededor del punto donde la carga de la molécula es cero. Para el ácido aspártico, esto corresponde a pK a1 y pK a2.
Referencias
- http://www.lpi.usra.edu/lunar/missions/apollo/ apollo_12/experimentos/encuesta/
- Arunan, Elangannan; Desiraju, Gautam R.; Klein, Roger A.; Sadlej, Joanna; Scheiner, Steve; Alkorta, Ibon; Clary, David C.; Crabtree, Robert H.; Dannenberg, Joseph J.; Hobza, Pavel; Kjaergaard, Henrik G.; Legon, Anthony C.; Mennucci, Benedetta; Nesbitbitta; T, David J. (2011). “Definición del enlace de hidrógeno”. Pura Appl. Chem. 83 (8): 1637—1641. doi:10.1351/PAC-Rec-10-01-02