
5.4: Buenas Prácticas de Documentación (PIB)
- Page ID
- 53069

Independientemente de las prácticas de orientación que se sigan, todas exhiben la misma filosofía de prácticas de documentación, a veces referidas como Buenas Prácticas de Documentación (GDP). La FDA utiliza el acrónimo ALCOA (atribuible, legible, contemporáneo, original y preciso) para describir la importancia de los GDP. La clave de ALCOA es una documentación exhaustiva para garantizar la reproducibilidad y trazabilidad.
La posición de la FDA (y la mayoría del sistema de calidad) con respecto a la documentación es, 'si no está escrita, no se hizo. ' La documentación adecuada es esencial en una empresa regulada desde el descubrimiento hasta las manos del cliente. Proporciona a los organismos reguladores, abogados, oficinas de patentes y editores de revisión por pares la información que necesitan para validar el proceso de fabricación del producto. Si bien las agencias reguladoras te dicen lo que debes hacer, no te dicen cómo. Las buenas prácticas actuales de fabricación (CGMP), por ejemplo, no son una receta para la fabricación de producción sino pautas. Este capítulo se centra en algunos elementos comunes e importantes de documentación que se encuentran tanto en lugares de trabajo regulados como no regulados.
La documentación sirve a tres propósitos fundamentales:
- Como herramienta de planeación de proyectos, la documentación mejora la comunicación de las metas y prioridades del proyecto.
- La documentación proporciona un registro histórico de quién-qué-cuando-por-qué-cómo; qué se hizo, cómo se hizo, qué se cambió, quién lo hizo, cuándo ocurrió y por qué se hizo. Los registros precisos suelen ser la mejor defensa de una firma en casos de litigio.
- Es requerido por las directrices y regulaciones CGMP, ISO, QSR y GLP que reconocen claramente que la documentación tiene buen sentido común.
La frase “documentación y trazabilidad” es familiar para todas las empresas que deben cumplir con las regulaciones de la FDA. Una empresa debe ser capaz de proporcionar registros para demostrar la trazabilidad de todas las partes de un producto terminado, incluyendo pero no limitado a materias primas, intermedios y lotes finales de lote. Un producto final puede ser liberado solo si la documentación que lo ha rastreado de principio a fin está completa, el producto ha cumplido con todas las especificaciones requeridas del producto y ha sido producido de acuerdo con la normativa necesaria. Por lo tanto, las empresas reguladas cuentan con sistemas que aseguran que se registre la obra, que se completen los documentos correspondientes y que los documentos se almacenen en un lugar seguro y fácilmente recuperable.
Los documentos que se archivan deben ser fácilmente recuperados en los casos en que los clientes cuestionen la calidad de un producto comprado, o cuando la empresa está siendo inspeccionada o auditada por una agencia reguladora. En todos los casos, las consecuencias de faltar documentación pueden ser graves. En efecto, la supervivencia misma de la compañía depende de estos documentos. La documentación es probablemente el primer y más significativo requisito de CGMP necesario en una nueva empresa de biotecnología. El reto es establecer, con recursos limitados y con un pequeño personal que pueda tener poca experiencia en CGMP, el mismo grado de cumplimiento de CGMP que las compañías farmacéuticas más grandes.
En algunas empresas, a medida que se desarrolla un nuevo producto para la producción, el proceso de mantenimiento de registros a menudo se ve como un inhibidor del avance del proyecto. La documentación ralentiza y carga las operaciones diarias debido al tiempo dedicado a llenar y firmar los formularios y luego archivarlos cuidadosamente. No hay atajo, sin embargo. Aunque cortar esquinas en el mantenimiento de registros puede parecer ventajoso al principio, la falta de documentación puede causar retrasos al hacer que experimentos y procesos fundamentales se repitan innecesariamente o resultar en conclusiones defectuosas. Cuando una empresa fabrica un farmacéutico, produce dos productos: el medicamento y los documentos anexos que se utilizaron para elaborar el medicamento.
En resumen, la documentación funciona para: Registrar lo que se ha hecho, establecer la propiedad, proporcionar a los trabajadores instrucciones específicas sobre cómo realizar una tarea, desarrollar especificaciones de producto, demostrar que el procedimiento se realizó correctamente, registrar parámetros experimentales, proporcionar una evidencia trail, asegura la trazabilidad, establece un contrato entre una empresa y un consumidor y establece un acuerdo entre una empresa y las agencias reguladoras.
Tipos de Documentación
La documentación es esencial en todas las áreas de trabajo de biotecnología, aunque los tipos específicos de documentos y los sistemas de documentación varían según el tipo de lugar de trabajo. Cada empresa contará con un conjunto de documentos para reflejar sus necesidades y requerimientos. Hay tres clases amplias de documentos.
- Los documentos de directiva instruyen a los empleados sobre cómo realizar una tarea. Los ejemplos incluyen procedimientos operativos estándar y protocolos.
- Los documentos de recolección de datos registran datos para proporcionar evidencia de que el documento directivo se realizó y se realizó correctamente.
- Los documentos de compromiso establecen el sistema de calidad de la organización; metas y estándares que se comprometen a seguir. Las declaraciones de misión, las declaraciones de visión y las declaraciones de calidad son ejemplos de documentos de compromiso.
Cuadernos de Laboratorio
Esta documentación permite a los investigadores reconstruir su trabajo, resolver problemas, detectar errores y demostrar a la comunidad científica que sus resultados fueron obtenidos adecuadamente y fueron reportados con precisión. Los cuadernos de laboratorio se pueden utilizar para establecer una reclamación de patente, asignar crédito para un descubrimiento original, documentar la integridad de los datos para su publicación y solucionar problemas. Es, por tanto, esencial que esté escrito con tinta indeleble y sea legible, claro y completo. Los datos de laboratorio pueden ser citados en litigios. Puede ser examinado por cualquier organismo regulador que lo solicite. La integridad de los portátiles es importante incluso en laboratorios de investigación no regulados.
Un cuaderno puede ser usado para documentar datos para apoyar publicaciones de investigación que han utilizado fondos gubernamentales, pueden apoyar una solicitud de patente, o pueden apoyar una nueva aplicación de medicamento en investigación o una nueva aplicación de medicamento a la FDA. Un cuaderno de laboratorio desordenado, o uno que no se mantenga con integridad, puede resultar en la pérdida de una patente, que le retiren los fondos de la subvención, tenga que pagar los fondos de la subvención, tener que pagar multas, perder su trabajo y recibir libertad condicional y tiempo en la cárcel.
¡Explora!
Lea el siguiente artículo de noticias sobre la mala conducta científica de un científico del VIH. ¿De qué se le acusa al Dr. Han? ¿Cuáles son algunos de los 'errores' que cometió que fácilmente podría haber evitado? ¿Cuáles son las ramificaciones que enfrenta de ser declarado culpable de mala conducta científica?
www.desmoinesregister.com/story/news/crime-and-courts/2015/07/01/dong-pyou-hansentencing-iowa-estado-científico-aids-vacuna-caso/29560297/
Conoce más aquí: https://en.Wikipedia.org/wiki/Scientific_misconduct
Procedimientos Operativos Estándar (SOP)
Las personas en instalaciones de producción utilizan documentos distintos a los cuadernos de laboratorio. Los Procedimientos Operativos Estándar (SOP) que describen cómo realizar una tarea son esenciales en las instalaciones de producción. Un procedimiento es un documento escrito que proporciona un esquema paso a paso de cómo se realiza una tarea. La mayoría de las instalaciones de producción (y muchos laboratorios) utilizan procedimientos para instruir al personal sobre cómo realizar procedimientos o tareas. Todos siguen los mismos procedimientos para garantizar que las tareas se realicen de manera consistente y correcta. Los SOP deben escribirse de manera que sean claros, fáciles de seguir y puedan acomodar cambios menores en la instrumentación. Los SOP suelen escribirse en oraciones de comando en lugar de en una narrativa. La colocación y distribución de los SOP son controlados y documentados, y se revisan periódicamente.
Los Procedimientos Operativos Estándar describen qué se requiere para realizar una tarea, qué problemas pueden surgir y cómo tratarlos, cómo documentar que la tarea se realizó correctamente y, por último, quién está calificado o responsable del trabajo.
Los SOP son importantes por muchas razones
- Proporcionar consistencia cada vez que se realiza un procedimiento o proceso.
- Servir como recordatorios para asegurar que el trabajo se realiza correctamente
- Se utiliza para formar a los nuevos empleados de la forma correcta de realizar el trabajo
- Reducir la posibilidad de falla al permitir que el empleado complete cualquier tarea
Formularios
Las formas a menudo se asocian con SOP. Estos formularios requieren que un individuo realice la tarea para monitorear el proceso o procedimiento a medida que se realiza. Rellenar los espacios en blanco e inicializar los pasos a medida que avanzan asegura que los pasos se sigan correctamente. En la producción, el formulario a menudo tiene espacios en blanco para registrar información sobre ID/números de lote de materias primas, pesos, tiempos, temperaturas y otra información necesaria para el control de calidad del producto final. En algunos laboratorios de producción, un testigo debe firmar pasos clave.
Protocolos
El término protocolo puede ser utilizado para referirse a un procedimiento que se realizará una sola vez y puede aplicarse a una tarea o experimento que tenga por objeto responder a una pregunta o probar una hipótesis. El protocolo describe los pasos que se deben seguir para realizar el experimento. Los SOP no pretenden dar lugar a la respuesta a una pregunta o probar una hipótesis. Los protocolos en las preguntas de investigación se abordan de manera continua. En instalaciones de producción, temas relacionados con el desempeño del producto, efectos del almacenamiento (tanto a corto como a largo plazo) en el producto, calidad del producto bajo diferentes condiciones, etc.
Informes
Un informe es un documento formal que describe los resultados de una tarea completada. El reporte resume lo que se hizo, por quién, por qué, los datos (resultados) y las conclusiones. Un reporte está escrito en una narrativa dirigida a un tipo particular de lector, con suficientes antecedentes e información técnica para lograr una cantidad adecuada de información. Por ejemplo, los informes a la administración de nivel superior pueden no incluir tantos detalles específicos como los informes dirigidos a los reguladores. Algunos informes se publican en revistas científicas, como los informes de investigación científica básica. Otros informes, como los de investigaciones realizadas en una empresa, podrán o no publicarse, pero deberán ponerse a disposición de los inspectores.
¡Explora!
Conoce la oficina de integridad de la investigación (ORI). ¿Qué hacen? Ir a: http://ori.hhs.gov/case_summary. Escoge un caso que te parezca interesante y resume los hallazgos del ORI y el castigo.
Los informes de laboratorio y los trabajos científicos tienen cuatro funciones típicas
- Para persuadir a otras personas para que acepten tu hipótesis con base en los datos que has presentado.
- Publicar sus datos, métodos, material y resultados para otros investigadores
- Convertirse en una parte aceptada de la comunidad científica contribuyendo al cuerpo de conocimiento
- Proporcionar un registro de investigación para documentación, almacenamiento y referencia futura
Libros de bitácora
Los cuadernos de registro se utilizan para mantener información sobre el estado y el mantenimiento de equipos o instrumentos. Los libros de registro suelen ser cuadernos encuadernados Cada vez que se utiliza un instrumento o equipo, se calibra, se realiza un mantenimiento preventivo y se repara el instrumento o artículo, esa información se registra en el cuaderno.
Documentos de Laboratorio Analítico
Los documentos analíticos de laboratorio contienen datos de pruebas analíticas que miden algunos parámetros en una muestra. Los laboratorios clínicos analizan la sangre en busca de componentes celulares, iones, fármacos y niveles enzimáticos. El producto es el resultado de la prueba. La documentación incluye la muestra que se está probando y la metodología de prueba. Los elementos de un documento analítico de laboratorio difieren de un laboratorio a otro y dependen en gran medida de las regulaciones, como CLIA (¡más sobre esto en un capítulo posterior!).
Sistemas de numeración
Los números de identificación se utilizan para identificar los elementos de manera única. Los números de identificación se utilizan con fines de trazabilidad y se utilizan para el inventario generalizado; ¡materias primas, productos, equipos e incluso documentos! Los números de identificación deben identificar el artículo de forma única.
Etiquetas
Las etiquetas identifican instrumentos, materias primas, productos u otros artículos. El formato y el contenido de las etiquetas están altamente regulados por la FDA. ¿Puedes encontrar el CFR para etiquetar medicamentos?
Formularios de Cadena de Custodia
Cadena de custodia es un término que se refiere al mantenimiento de un registro ininterrumpido de posesión de una muestra desde el momento en que se recolecta a través de la entrega, recepción, almacenamiento, análisis o disposición. Los documentos de cadena de custodia son un método para organizar la información sobre muestras. El establecimiento de procedimientos de cadena de custodia es necesario porque los resultados de las pruebas o análisis se pueden sostener como prueba en los procesos de litigio. A cada muestra se le asigna un número de identificación único y se registra la entrada y salida, a medida que se procesa. Para demostrar la importancia de las formas de cadena de custodia, considere, el veredicto del juicio de O.J. Simpson se basó en documentación inadecuada de la cadena de custodia de las pruebas de ADN. Si bien la ciencia de las huellas dactilares del ADN era sólida y rigurosa, la mala documentación de quién manejaba las muestras de sangre, cuándo, dónde y cómo, llevó a la absolución.
Registros de entrenamiento
La FDA requiere un programa de capacitación continua documentado para el cumplimiento de las regulaciones CGMP. Es responsabilidad del Aseguramiento de la Calidad verificar que se implemente un programa de capacitación CGMP y que sea un programa continuo. Además de la capacitación de CGMP, la normativa exige que todos los empleados estén adecuadamente capacitados en sus funciones laborales, ya sean nuevos empleados o empleados existentes que estén aprendiendo nuevas metodologías o la operación de nuevos equipos. La capacitación se basa en los SOP escritos y aprobados propios de la compañía. Debe estar bien documentado y proporcionar las herramientas y conocimientos necesarios para capacitar a los empleados.
Presentaciones reglamentarias
Las presentaciones reglamentarias son documentos diseñados para cumplir con los requisitos de una agencia reguladora externa. Las compañías farmacéuticas deben presentar una solicitud ante la FDA, mostrando su investigación preliminar sobre un medicamento, su plan para ensayos clínicos y otra información relevante antes de que puedan comenzar a probar en campo un nuevo medicamento en humanos.
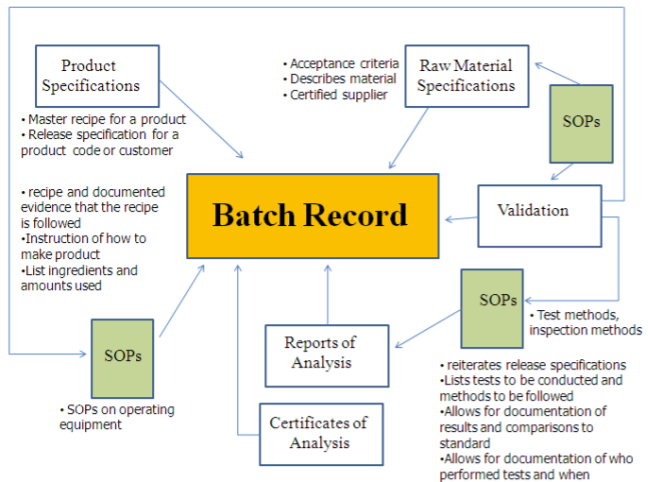
Registros por lotes (BPR)
Los registros de producción por lotes (BPR) son un requisito de las Buenas Prácticas de Manufactura. Se trata de una copia exacta del Registro Maestro de Producción y Control correspondiente. Los BPR están cuidadosamente diseñados para que toda la información apropiada del proceso sea documentada y demostrada por escrito. Los BPR deben ser revisados para verificar su exactitud y deben ser firmados y fechados por un grupo de calidad antes de su uso en la fabricación. Un BPR es una combinación de un documento SOP y un formulario en el sentido de que dirige a los operadores en cómo hacer el producto y cada paso tiene espacios en blanco que se llenan a medida que el técnico realiza la acción.
Para los pasos críticos, se requiere que un testigo vigile y firme el BPR. Los registros por lotes son documentos legales y forman parte del cumplimiento de la validación del proceso. El departamento de calidad emite oficialmente el registro del lote al equipo de producción, y es esencial que los espacios en blanco se llenen a medida que se realizan los procedimientos. Los BPR pueden estar en una ubicación central o distribuidos en diferentes áreas siempre que sean fácilmente recuperados y archivados de manera lógica y ordenada. Los BPR deben conservarse por un mínimo de 1 año después de la fecha de vencimiento de un lote correspondiente del producto.
Documentación electrónica
El campo de la biotecnología utiliza una mezcla diversa y compleja de documentación en papel y electrónica. Hay muchas ventajas e inconvenientes para ambos, pero las empresas tienden a elegir el proceso de documentación que mejor se adapte a sus necesidades y al mismo tiempo cumplir con los requisitos reglamentarios. Ante el uso extensivo de la documentación electrónica y la demanda de regulación sistemática de dicha documentación, en 1997, la FDA emitió el reglamento 21 CFR Parte 11 Firmas Electrónicas; Regla Final, para atender estas preocupaciones. En 2003, la FDA publicó su “Guía para la Industria Parte 11, Registros Electrónicos; Firmas Electrónicas — Alcance y Aplicación”.
El propósito de estas regulaciones es incentivar a las compañías farmacéuticas a adoptar métodos modernos de documentación electrónica, requiriendo que validen estos métodos electrónicos como seguros, confiables y tan buscables como las prácticas de documentación en papel. En la siguiente tabla se esboza un vocabulario utilizado en relación con 21 CFR Parte 11.
21 CFR Parte 11
- Rastro de Auditoría. Sendero de marca de tiempo generado por computadora.
- Biometría. Método para identificar la identidad de un individuo.
- Sistema Cerrado. Solo accesible para personas que necesitan el sistema.
- Cuaderno Electrónico de Laboratorio. Programas de software de computadora diseñados para su uso como cuaderno de laboratorio.
- Registros Electrónicos. Texto, gráficos, datos, información de audio que se crea modificada, mantenida, archivada, recuperada o distribuida por un sistema informático.
- Firma Electrónica. Equivalente a una firma manuscrita.
- Software de encriptación. Traduce la información en un código secreto
- Sistema Híbrido. Utiliza ambos sistemas; papel y electrónico.
- Sistema de Gestión de la Información de Laboratorio (LIMS). Sistema de gestión de laboratorio basado en computadora
¡Explora!
Lea el siguiente artículo sobre mala conducta científica no intencional en el uso de cuadernos de laboratorio de papel. De lo que aprendiste en este artículo, ¿cuáles son algunos ejemplos de mala conducta científica no intencional? ¿Cómo puede un cuaderno electrónico ayudar a evitar algunas trampas de mala conducta científica usando cuadernos electrónicos?
Gestión del cambio en la documentación
Es necesario que una instalación de producción siga los mismos procedimientos al pie de la letra con cada lote de producto producido, y es importante que todos los análisis de laboratorio de soporte también apoyen un solo conjunto de protocolos para producir de manera confiable un resultado consistente. Esta rígida adherencia a procedimientos cuidadosamente descritos ayuda a prevenir resultados inconsistentes, pero sofoca mejoras que podrían ayudar a mejorar o agilizar un proceso. Cuando se realiza un cambio, el cambio es acordado cuidadosamente por todas las partes involucradas, y todos los involucrados deben tener un procedimiento para promulgar el cambio.
Por lo general, cualquier persona afectada por el cambio propuesto puede hacer una solicitud de cambios en los métodos, hojas de datos de muestreo o instrucciones de calibración. La solicitud se realiza por escrito siguiendo el procedimiento de cambio establecido por la empresa. Un comité o Gerente de Aseguramiento de Calidad generalmente aprueba dicha solicitud dependiendo de la estructura jerárquica de la empresa, y el cambio que se esté realizando. El departamento de Aseguramiento de la Calidad suele ser responsable de que todas las copias de documentos obsoletos se archiven electrónicamente, y las copias impresas sean retiradas y destruidas. También son responsables de monitorear la actividad para asegurar que los cambios aprobados se incorporen a la actividad laboral rutinaria del laboratorio, y el cambio no ha tenido efectos nocivos conocidos.
Almacenamiento y recuperación de documentos
Es crucial que todos los archivos relacionados con la fabricación de productos se mantengan de forma segura y se pueda acceder fácilmente durante las inspecciones del sistema de calidad (por ejemplo, por los auditores de la FDA o ISO). Todos los registros, cuando no estén en uso, deben conservarse en trasteros cerrados (e ignífugos). La seguridad de los datos informáticos se mantendrá mediante procedimientos operativos estándar de procesamiento de datos para restringir la entrada a la información de datos informáticos.
El tiempo que se mantienen los registros está determinado por las regulaciones, el producto fabricado y las políticas de la compañía. Los registros originados y mantenidos como copias impresas se conservan durante cinco años, y tres años después de que se haya publicado el lote. Muchas empresas han pasado a transferir papel a formato electrónico después de cinco años y se han mantenido indefinidamente. Los registros generados por computadora se conservarán en esa forma indefinidamente, con el debido cuidado para mantenerlos en un almacenamiento seguro y libre de riesgos.
En resumen, todos los materiales recibidos y utilizados, y todos los procedimientos y procesos seguidos por una firma son cuidadosamente descritos y seguidos, dejando un rastro de papel que se archiva cuidadosamente. Sin embargo, esto puede ser gravoso para la empresa; es esencial en el aseguramiento de la calidad y para proteger a la firma de litigios. Además, este rastro de papel es requerido por muchas agencias reguladoras y es clave para acceder a los mercados internacionales.
Reglamento de documentos
Dependiendo del sistema de calidad utilizado (y las regulaciones que rodean ese sistema de calidad), existen muchos tipos diferentes de requisitos de documentación del sistema de calidad. A continuación se presenta una breve revisión de las regulaciones del sistema de calidad en los sistemas CGMP e ISO 9000.
21 CFR 211
El Título 21 El Capítulo 1 del CFR contiene todas las regulaciones relativas a la producción segura de alimentos, medicamentos, dispositivos médicos, diagnósticos y productos biológicos para uso humano y animal bajo la supervisión de la FDA. (fda.gov) La documentación se encuentra bajo la Parte 211.
¡Explora!
Vaya a la base de datos CFR y busque la Parte 211.
¿Qué subparte cubre los registros? ¿Cuánto tiempo hay que mantener los registros? Describa brevemente cada una de las secciones de Registros e Informes de 8 subpartes.
Documentación ISO 9001
Un sistema de calidad válido, incluso voluntario como ISO 9001, requiere una documentación rigurosa y un mantenimiento disciplinado de registros. Algunas de las actividades de mantenimiento de registros requeridas por la ISO 9000 incluyen, pero no se limitan a, registros de capacitación, políticas, procedimientos, instrucciones, protocolos, registros de compras, datos de pruebas, registros de auditoría y registros de calibración. Este tipo de documentación forma parte de la prueba requerida para demostrar que la empresa está siguiendo los lineamientos ISO en su sistema de gestión de calidad y como tal, es un componente necesario para mantener su acreditación.
Los procesos de documentación en una organización pueden diferir según el tamaño de la organización, el alcance y la complejidad de sus actividades y muchos otros factores. No obstante, una cosa es segura, para la certificación ISO 9001, esta documentación debe ser minuciosa, completa y actualizada. La guía ISO 9001:2015 sobre documentación permite flexibilidad organizativa en la forma que elige para documentar su sistema de gestión de calidad (SGC). ISO permite a cada empresa determinar qué documentación es necesaria para acreditar la planeación efectiva, operación y control de sus procesos, y la mejora continua de la efectividad de su SGC. ISO 9001:2015 cláusula 4.4 Los sistemas de gestión de la calidad y sus procesos requieren que una organización “mantenga la información documentada en la medida necesaria para apoyar la operación de los procesos y retener la información documentada en la medida necesaria para tener confianza en que los procesos son llevado a cabo según lo previsto.” (ISO, 2015). La guía sobre el requisito de información documentada para ISO se puede encontrar aquí: http://www.iso.org/iso/documented_information.pdf
¡Pon a prueba tus conocimientos!
Como Gerente de Calidad de Proteínas 'R Us, usted supervisa el Departamento de Calidad de una firma farmacéutica y ha sido informado por el abogado de un cliente de que su cliente ha sufrido daños severos por su producto antibiótico y tiene la intención de demandar por daños. Explique de qué manera cada uno de los siguientes documentos podría desempeñar un papel en la protección de su firma de este litigio:
- Cuadernos de laboratorio de I+D
- Informes mensuales desde I+D hasta administración de nivel superior
- Libros de registro de equipos de producción
- Etiqueta del producto
- Formularios de cadena de custodia
- Registros de capacitación de personal de producción
- BPRs