
6.3: Diferentes procesos de revisión de aplicaciones de medicamentos
- Page ID
- 53258

Aplicación de nuevos medicamentos (NDA)
Si el medicamento pasa las tres fases de prueba, la compañía puede presentar una Solicitud de Nuevo Medicamento (NDA), que permite a la FDA determinar que el nuevo medicamento (o biológico) es seguro, confiable y efectivo para las indicaciones en el etiquetado. La FDA emplea revisores expertos que examinan los resultados de las pruebas y determinan si el nuevo medicamento puede ser aprobado.
Tipos de Aplicaciones de Medicamentos:
- Tradicional 505 (b) (1) NDA — Ley FD&C, artículo 505 (b) (1)
- 505 (b) (2) NDA
- NDA abreviado (ANDA) 505 (j)
- Original BLA — Ley PHS, Sección 351 (a) — para los biológicos discutidos en el siguiente capítulo
- Biosimilar BLA — Ley PHS, Sección 351 (k) — para biosimilares discutidos en el siguiente capítulo
Nueva Aplicación del Mismo Medicamento - Proceso de Revisión
Aprobación de una nueva aplicación del mismo medicamento: Si una empresa quiere ampliar el uso de su medicamento a una nueva aplicación, los estudios previos de seguridad suelen seguir siendo aplicables; sin embargo, se requerirán nuevos estudios clínicos para probar la eficacia del medicamento para su nueva aplicación. El 505 (b) (1) NDA es la aplicación completa con toda la información del estudio adecuada esbozada en el CFR que demostrará la seguridad y efectividad del medicamento. El 505 (b) (2) NDA puede incluir medicamentos donde la seguridad y la efectividad se han establecido en estudios previos (de otras empresas), permitiendo a las empresas desarrollar tratamientos más rápido con menos voluntarios de estudio clínico. Un ejemplo de una aplicación sería en lugar de un tratamiento de diez días del medicamento, el medicamento ahora tiene una cápsula de liberación lenta, por lo que solo tiene tratamiento de 3 días, pero se libera lentamente a lo largo de diez días.
Aplicación abreviada de nuevos medicamentos (ANDA)
Curiosamente, el término 'medicamento genérico' no está definido en las regulaciones de la FDA. Un medicamento genérico debe tener el mismo ingrediente activo, la misma potencia y la misma dosis para ser vendido sin tener que repetir los extensos ensayos clínicos utilizados en el desarrollo de los medicamentos originales de marca. Los genéricos deben entregar la misma cantidad de ingrediente activo en el torrente sanguíneo de un paciente durante el mismo período que la marca, esto se conoce como bioequivalencia. La velocidad y el grado de absorción de un fármaco se denominan su biodisponibilidad. Si la biodisponibilidad de los dos es similar, los fármacos son bioequivalentes.
Un ANDA (505 (j)), es una solicitud de aprobación para un producto farmacológico bioequivalente. Esta solicitud contiene datos enviados al Centro de Evaluación e Investigación de Medicamentos de la FDA, Oficina de Medicamentos Genéricos para su revisión para su aprobación. Las aplicaciones genéricas de medicamentos se denominan “abreviadas” porque con frecuencia no se requiere que incluyan datos preclínicos (animales) o clínicos (humanos) para mostrar eficacia y seguridad. En cambio, un solicitante genérico debe demostrar científicamente que su producto tiene equivalencia terapéutica. Equivalencia terapéutica significa que el medicamento debe tener el mismo efecto clínico y seguridad (bajo el mismo etiquetado) que el medicamento no genérico y debe tener el mismo ingrediente activo, con idéntica fuerza, calidad, pureza y potencia. No necesita tener los mismos ingredientes inactivos. Una vez aprobado, la compañía puede entonces fabricar y comercializar el medicamento genérico para proporcionar una alternativa de bajo costo al medicamento de marca.
Solicitud de Licencia Biológica (BLA)
Se requiere BLA para productos biológicos sometidos a CBER o CDER (proteína caracterizada). El BLA debe incluir toda la información de seguridad y eficacia necesaria para la aprobación del medicamento. Una solicitud 351 (a) (Original BLA), contiene toda la información requerida y esbozada en 21 CFR 601.2. Una aplicación 351 (k) es una BLA abreviada para un biosimilar. Aunque algunos productos biológicos son supervisados por CDER, el proceso BLA se explora más a fondo en el capítulo de Aprobación Regulatoria de Biológicos.
Revisiones aceleradas
La FDA identifica cuatro procesos de aprobación acelerados: vía rápida, terapia innovadora, revisión prioritaria y aprobación acelerada. Estos procesos acelerados pueden ayudar a que los medicamentos lleguen a manos de los consumidores más rápido en función de sus necesidades tangibles, como tratar una enfermedad grave con un nuevo medicamento o uno que se mejore sustancialmente con respecto a las terapias actuales. Obtenga más información aquí: www.fda.gov/patients/learnabout-drugs and-device-approvals/fast-track-breakthrough-therapy-accelerated-approval-priority-review
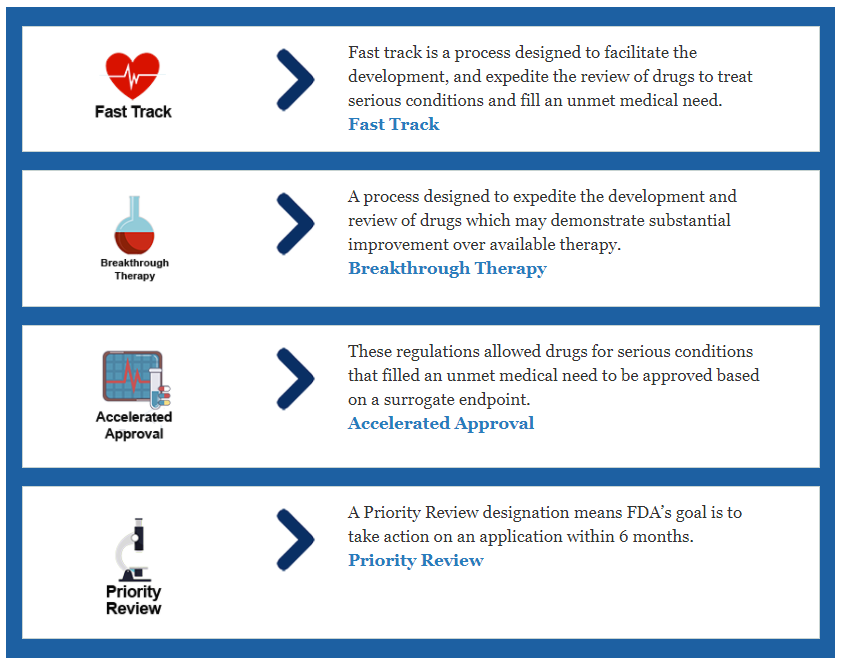
- Fast Track es un proceso diseñado para facilitar el desarrollo y agilizar la revisión de medicamentos para tratar afecciones graves y cubrir una necesidad médica insatisfecha para asegurar nuevos medicamentos vitales al paciente antes. Fast Track aborda una amplia gama de afecciones graves.
- La designación de terapia innovadora es un proceso diseñado para acelerar el desarrollo y revisión de medicamentos que están destinados a tratar una afección grave, y la evidencia clínica preliminar indica que el medicamento puede demostrar una mejoría sustancial sobre la terapia disponible en un punto (s) final (s).
- Revisiones Prioritarias Estos productos representan mejoras significativas en la seguridad o eficacia del tratamiento de un padecimiento grave en comparación con los productos comercializados actualmente.
- Aprobación acelerada Este programa garantiza que los productos para afecciones graves estén disponibles más temprano en el proceso de desarrollo al confiar en un criterio de valoración sustituto que puede predecir un beneficio clínico, como una contracción tumoral puede significar un aumento de la supervivencia.
Programa Especial de Incentivos a
Existen varios programas patrocinados para incentivar el desarrollo de un medicamento que está en demanda. La Ley de Medicamentos Huérfanos y GAIN (Generando Incentivos Antibióticos Ahora) y el Plan Presidencial de Emergencia para el Alivio del Sida (PEPFAR) son tres ejemplos de programas especiales de incentivos GAIN es un programa de incentivos para desarrollar medicamentos que traten infecciones bacterianas potencialmente mortales. Los medicamentos GAIN obtienen revisión Fast Track y Priority además de exclusividad de mercado a 5 años (¡7 años. para medicamentos huérfanos!). Para obtener más información sobre la Ley de Seguridad e Innovación de la FDA, visite: www.fda.gov/regulatoryinformation/lawsenforcedbyfda/significantamendmentstothefdcact/f DASIA/ucm20027187.htm
La FDA sí permite la aprobación de medicamentos y vacunas destinados a contrarrestar el terrorismo biológico, químico y nuclear sin probar primero su seguridad y valor en los ensayos de Fase II y III. Sería poco ético exponer deliberadamente a los humanos a radiaciones dañinas o patógenos para probar la efectividad del tratamiento. Por ejemplo, la FDA aceleró la aprobación de un nuevo medicamento, Cipro ®, un antibiótico que trata adecuadamente a quienes están expuestos al ántrax. La FDA también tiene un proceso más simplificado para la aprobación de “medicamentos huérfanos” (medicamentos con un pequeño número de beneficiarios pero con gran beneficio).
Medicamentos Huérfanos
La FDA administra un programa que brinda incentivos para desarrollar medicamentos para su uso en poblaciones de pacientes de enfermedades raras (200,000 o menos casos), o si existe una expectativa razonable de que el medicamento no se desarrollará sin la asistencia de la FDA. Para conocer más sobre las enfermedades raras, consulte la Oficina de Investigación de Enfermedades Raras: http://rarediseases.info.nih.gov/. Las empresas que fabrican medicamentos huérfanos reciben los siguientes incentivos: exclusividad de comercialización a siete años, un crédito fiscal para la investigación clínica asociada al producto, asistencia de diseño de investigación de la FDA y subvenciones
¡Pon a prueba tus conocimientos!
Descargue y lea Estudio de Caso 1: Aprobación de Medicamentos: Traer un Nuevo Medicamento al Mercado. Revisaremos este caso de estudio https://www.fda.gov/media/94428/download
- En el caso de estudio, el Dr. Green ha contratado al experto en asuntos regulatorios Dr. Robert's, para guiar el desarrollo de la compañía y la aprobación por la FDA de Lowagliflozin, un NME para tratar la diabetes tipo 2. ¿Qué es un NME y por qué Lowagliflozin califica como NME?
- Explique dónde está Lowagliflozina en el proceso de aprobación y los hitos restantes por recorrer.
- El Dr. Green pregunta al consultor si es posible hacer una revisión expedita para Lowagliflozina. ¿Qué es una revisión acelerada? ¿Cuáles son los cuatro programas de revisión acelerada que tiene la FDA? ¿Cuáles crees que son las posibilidades de obtener una revisión expedita?
- Una vez que Lowagliflozina obtiene la aprobación de la FDA, ¿ese es el final de su interacción con la FDA? Explique.
Proceso de revisión de medicamentos de venta libre (OTC)
Los medicamentos de venta libre (OTC) son medicamentos que se pueden obtener y usar sin receta médica. Más de 300,000 medicamentos de venta libre están disponibles en Estados Unidos. Los medicamentos de venta libre siguen siendo supervisados por CDER para garantizar que estén debidamente etiquetados, y los beneficios de su uso superan los riesgos. Para ser designados OTC, los medicamentos deben considerarse seguros, los beneficios superan los riesgos y tienen un potencial mínimo de abuso.
Los medicamentos de venta libre aún pueden conllevar riesgos, en particular, riesgos de efectos secundarios, interacciones medicamentosas o sobredosis. Un ejemplo es Tylenol (ingrediente activo acetaminofén), que la mayoría de la gente asume que es muy seguro. Sin embargo, una sobredosis de este medicamento de venta libre resulta en más de 44 mil individuos en la sala de emergencias y más de 400 mueren cada año de insuficiencia hepática. ¡Conoce más aquí! https://www.sciencedaily.com/releases/2015/06/150622124713.htm
Detrás del mostrador
Algunos medicamentos de venta libre se mantienen técnicamente “detrás del mostrador” como el anticonceptivo de emergencia y la pseudoefedrina. Se impide intencionalmente a los clientes el acceso directo a estos medicamentos debido a su riesgo de uso indebido involuntario o intencional, como ocurre con algunos medicamentos para el resfriado que contienen pseudoefedrina. Aunque no se requiere receta para pseudoefedrina, el farmacéutico solo puede prescindir de la edad y la verificación de la aplicación.
Desarrollo y revisión de medicamentos de venta libre
Aunque muchos medicamentos están aprobados para su uso de venta libre a través del proceso de revisión de la nueva aplicación de medicamentos (OTC NDA), otros medicamentos de venta libre están regulados bajo la Monografía OTC. Este proceso se basa en monografías publicadas, que describen ingredientes aceptables, dosis, formulaciones y etiquetado de consumo para medicamentos de venta libre. Los productos que se ajustan a una monografía final, y son generalmente reconocidos como seguros y efectivos (GRASE), pueden comercializarse sin autorización previa de la FDA.
Los medicamentos que no son GRASE pueden estar sujetos a aprobación previa de la FDA a través de un NDA ya sea a través de un NDA directo a OTC o cambio de designación de receta a venta libre. Una NDA directa a OTC requerirá el mismo proceso de aprobación de medicamentos que un medicamento recetado, excepto que puede necesitar una población más amplia de pruebas clínicas. El medicamento de venta libre para el dolor frío Abreva (docosanol) es un ejemplo de una aplicación directa a OTC. Una ruta más común es el switch Prescription-to-OTC después de que expira una patente. Esta ruta es popular debido a la Ley Hatch-Waxman, que califica a las empresas para una extensión de 3 años de exclusividad de patentes. Una ruta de prescripción a OTC requiere una demostración de que la necesidad de supervisión médica es innecesaria; el paciente puede autodiagnosticarse y el producto tiene baja toxicidad.
En resumen, todos los medicamentos de venta libre deben cumplir con los mismos requisitos reglamentarios y regulaciones de cGMP para productos farmacéuticos, como se describe en 21 CFR 210 y 211. Los sitios de fabricación también deben registrarse ante la FDA, estar sujetos a inspecciones de rutina y estar en la lista de medicamentos. Sin embargo, los medicamentos de venta libre se someten a un escrutinio adicional para garantizar que los consumidores puedan usarlos de manera segura y efectiva sin la supervisión del proveedor médico.