
4.1: Introducción
- Page ID
- 54793

Un tema recurrente de este trabajo es adoptar un enfoque computacional global para analizar elementos de genes y ARN codificados en el genoma y utilizarlo para encontrar nuevos fenómenos biológicos interesantes. Podemos hacer esto viendo cómo los ejemplos individuales “divergen” o difieren del caso promedio. Por ejemplo, al examinar muchos genes que codifican proteínas, podemos identificar características representativas de esa clase de loci. Luego podemos elaborar pruebas de alta precisión para distinguir genes que codifican proteínas de genes que no codifican proteínas. A menudo, estas pruebas computacionales, basadas en miles de ejemplos, serán mucho más definitivas que las pruebas convencionales de laboratorio húmedo de bajo rendimiento. (Tales pruebas pueden incluir espectrometría de masas para detectar productos proteicos, en los casos en los que queremos saber si un locus en particular es codificante de proteínas).
Motivación y Desafío
A medida que el costo de la secuenciación genómica continúa bajando, la disponibilidad de datos genómicos secuenciados se ha disparado. Sin embargo, el análisis de los datos no se ha mantenido al día, mientras que hay muchos fenómenos biológicos interesantes que no se han descubierto en las interminables cadenas de ATGCs. El objetivo de la genómica comparativa es aprovechar la gran cantidad de información disponible para buscar patrones biológicos.
Como su nombre indica, la genómica comparada no se enfoca en un conjunto específico de genomas. El problema de centrarse puramente en el nivel del genoma único es que se pierden las firmas evolutivas clave. La genómica comparativa resuelve este problema comparando genomas de muchas especies que evolucionaron a partir de un ancestro común. A medida que la evolución cambia el genoma de una especie, deja huellas de su presencia. Veremos más adelante en este capítulo que la evolución discrimina entre porciones de un genoma sobre la base de la función biológica. Al explotar esta correlación entre las huellas dactilares evolutivas y el papel biológico de una subsecuencia genómica, la genómica comparativa es capaz de dirigir la investigación de laboratorio húmedo a partes interesantes del genoma y descubrir nuevos fenómenos biológicos.
FAQ
P: ¿Por qué las mutaciones solo se acumulan en ciertas regiones del genoma, mientras que otras regiones se conservan?
R: En regiones no funcionales del ADN, las mutaciones acumuladas se mantienen porque no perturban la función del ADN. En las regiones funcionales, estas mutaciones pueden conducir a una disminución de la aptitud; estas mutaciones luego se descartan de la especie por selección natural.
Podemos obtener mucha información sobre la evolución a través del estudio de la genómica y, de manera similar, podemos aprender sobre el genoma a través del estudio de la evolución. Por ejemplo, a partir del principio de “supervivencia del más apto”, podemos comparar especies relacionadas para descubrir qué partes del genoma son elementos funcionales. El proceso evolutivo introduce mutaciones en cualquier genoma. En regiones no funcionales del ADN, las mutaciones acumuladas se mantienen porque no perturban la función del ADN. Sin embargo, en regiones funcionales, las mutaciones acumuladas a menudo conducen a una disminución de la aptitud Por lo tanto, no es probable que estas mutaciones decrecientes del estado físico se perpetúen a las generaciones futuras. A medida que avanza el tiempo, es probable que los organismos evolutivamente inadecuados no sobrevivan y sus genes se diluyen. Al comparar los genomas de las especies supervivientes con los genomas de sus antepasados, podemos ver qué porciones constituyen elementos funcionales y cuáles constituyen “ADN basura”.
A la fecha se han descubierto diversos marcadores y fenómenos biológicos importantes a través de métodos de genómica comparativa. Por ejemplo, CRISPRs (Clustered Regularmente Interspaced Short Palindromic Repeats), que se encuentran en bacterias y arqueas, se descubrieron por primera vez a través de la genómica comparativa. Los experimentos de seguimiento revelaron que proporcionan inmunidad adaptativa a plásmidos y fagos. Otro ejemplo, que veremos más adelante en este capítulo, es el fenómeno de la lectura de codones de parada, donde ocasionalmente se ignoran los codones de parada durante el proceso de la fase de traducción de la biosíntesis de proteínas. Sin una genómica comparada que los guíe, los experimentalistas podrían haber ignorado ambas características durante muchos años.
Sin un sistema para interpretar e identificar características importantes en los genomas, todas las secuencias de ADN en la tierra son solo un mar de datos sin sentido. Sin embargo, no podemos ignorar la importancia tanto de la informática como de la biología en la genómica comparada. Sin conocimiento de biología, uno podría pasar por alto las firmas de sustituciones sinónimos o mutaciones de cambio de marco. Por otro lado, ignorar los enfoques computacionales conduciría a la incapacidad de analizar conjuntos de datos cada vez más grandes que emergen de los centros de secuenciación. La genómica comparada requiere habilidades y conocimientos multidisciplinarios poco frecuentes.
Este es un momento particularmente emocionante para ingresar al campo de la genómica comparada, porque el campo es lo suficientemente maduro como para que existan herramientas y datos disponibles para hacer descubrimientos. Pero es lo suficientemente joven como para que probablemente se sigan haciendo hallazgos importantes durante muchos años.
Importancia de muchos genomas estrechamente relacionados
Para resolver características biológicas significativas necesitamos tanto similitud suficiente para permitir la comparación como suficiente divergencia para identificar firmas de cambio a lo largo del tiempo evolutivo. Esto es difícil de lograr en una comparación por pares. Mejoramos la resolución de nuestro análisis extendiendo el análisis a muchos genomas simultáneamente con algunos grupos de organismos similares y algunos organismos diferentes. Una simple analogía es la de observar una orquesta. Si colocas un solo micrófono, será difícil descifrar la señal proveniente de todo el sistema, ya que se verá abrumada por el ruido local desde el único punto de observación, el instrumento más cercano. Si colocas muchos micrófonos distribuidos por la orquesta a distancias razonables, entonces obtienes una perspectiva mucho mejor no solo de la señal general, sino también de la estructura del ruido local. Del mismo modo, al secuenciar muchos genomas a través del árbol de la vida, podemos distinguir las señales biológicas de los elementos funcionales del ruido de las mutaciones neutras. Esto se debe a que la naturaleza selecciona para la conservación de elementos funcionales a través de grandes distancias filogenéticas mientras constantemente introduce ruido a través de procesos mutagénicos que operan en escalas de tiempo más cortas
En este capítulo, asumiremos que ya tenemos una alineación genómica completa de múltiples especies estrechamente relacionadas, abarcando regiones codificantes y no codificantes. En la práctica, construir ensamblajes genómicos completos y alineaciones de genoma completo es un problema muy desafiante; ese será el tema del próximo capítulo.
FAQ
P: ¿Por qué hay más poder de resolución cuando aumenta la distancia evolutiva o la longitud de las ramas entre especies?
R: Si estamos comparando dos especies como el humano y el chimpancé que están muy cerca entre sí, esperamos ver pocas o ninguna mutación. Esto nos da poco poder discriminativo porque no vemos diferencia entre el número de mutaciones en los elementos funcionales frente al número de mutaciones en elementos no funcionales. Sin embargo, a medida que aumentamos el tiempo evolutivo entre especies, esperamos ver más mutaciones, pero lo que en realidad vemos es una disminución notable en el número observado de mutaciones en ciertas regiones del genoma. Podemos concluir que estas regiones son regiones funcionales. Por lo tanto, nuestra confianza en los elementos funcionales percibidos aumenta a medida que aumenta la longitud de
FAQ
P: ¿Por qué es mejor tener muchas especies estrechamente relacionadas para la misma longitud de rama en lugar de una especie lejanamente relacionada?
R: A medida que aumenta la longitud de las ramas entre especies distantes relacionadas, incluso los elementos funcionales no se conservan. Además, alinear de manera confiable genes de parientes lejanos de la misma especie es difícil, si no imposible, usando la tecnología actual como BLAST.
Genómica comparada y firmas evolutivas
Dado un alineamiento de todo el genoma, posteriormente podemos analizar el nivel de conservación de los elementos funcionales en cada uno de los genomas considerados. Usando el navegador del genoma UCSC, uno puede ver un nivel de conservación para cada gen en el genoma humano derivado de alinear los genomas de muchas otras especies. En la Figura 4.1 a continuación, vemos una secuencia de ADN representada en el eje x, mientras que cada “fila” representa una especie diferente. El eje y dentro de cada fila representa la cantidad de conservación para esa especie en esa parte del cromosoma (aunque también se utilizaron otras especies que no se muestran para calcular la conservación). Barras superiores corresponden con mayor conservación.
A partir de esta cifra, podemos ver que hay bloques de conservación separados por regiones que no están conservadas. Los 12 exones (resaltados por rectángulos rojos) se conservan en su mayoría entre especies, pero a veces faltan ciertos exones; por ejemplo, al pez cebra le falta el exón 9. Sin embargo, también vemos que hay un pico en algunas especies (como un círculo en rojo) que no corresponde a un gen codificante de proteínas conocido. Esto nos dice que algunas regiones intrónicas también se han conservado evolutivamente, ya que las regiones de ADN que no codifican proteínas aún pueden ser importantes como elementos funcionales, como ARN, microARN y motivos reguladores. Al observar cómo se conservan las regiones, en lugar de solo mirar la cantidad de conservación, podemos observar 'firmas evolutivas' de conservación para diferentes elementos funcionales.
El patrón de mutación/inserción/deleción puede ayudarnos a distinguir diferentes tipos de elementos funcionales en el genoma. Diferentes elementos funcionales están bajo diferentes presiones selectivas y al considerar a qué presiones selectivas se encuentra cada elemento, podemos desarrollar firmas evolutivas características de cada función. Por ejemplo, vemos la diferencia en las firmas evolutivas tal como muestran los genes codificantes de proteínas frente a los motivos reguladores... etc.
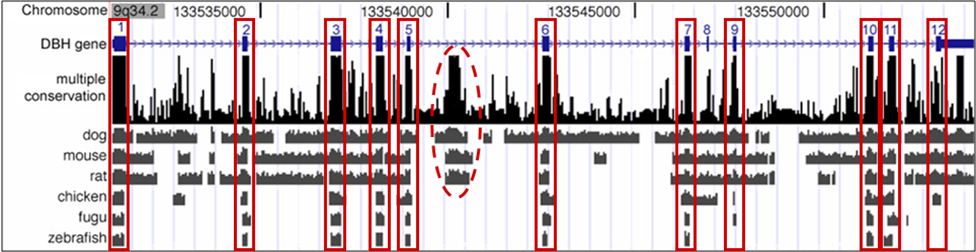
Figura 4.1: Los exones (en caja en rojo) están profundamente conservados de mamíferos a peces. Otros elementos también están fuertemente conservados, como el pico circular cerca del centro de la gráfica. Este puede ser un elemento regulador que se encuentra en mamíferos pero no en aves o peces.
FAQ
P: Dada una alineación de genes de múltiples especies, ¿qué se puede medir para determinar el nivel de conservación de uno o varios genes específicos?
R: Un método simple es solo observar la puntuación de alineación para cada gen. Si se quiere distinguir entre segmentos codificantes de proteínas altamente conservados de segmentos que no codifican proteínas, también se puede observar la conservación de codones. Sin embargo, en ambos enfoques, tenemos que considerar la posición de cada especie comparada en el árbol filogenético. Una puntuación de comparación por pares que sea menor entre dos especies separadas por una mayor distancia en el árbol filogenético que la puntuación por pares entre dos especies estrechamente relacionadas no implicaría necesariamente una menor conservación.