
4.3: Restricción en exceso
- Page ID
- 54776

En la mayoría de las regiones del genoma donde vemos conservación entre especies, esperamos que haya al menos alguna cantidad de sustitución sinónima. Se trata de sustituciones de nucleótidos “silenciosas” que modifican un codón de tal manera que el aminoácido que codifica no se modifica. En un artículo 2 de 2011, Lindblad—Toh et al. estudiaron la restricción evolutiva en el genoma humano haciendo análisis comparativo de 29 especies de mamíferos. Encontraron que entre los 29 genomas, el sitio nucleotídico promedio mostró 4.5 sustituciones por sitio.
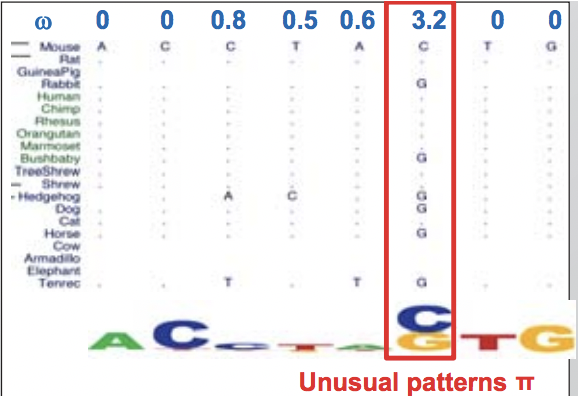
Figura 4.4: Esta secuencia muestra una tasa de sustitución inusual de sustituir C por G y viceversa.
Dada una tasa de sustitución promedio tan alta, no esperamos ver una conservación perfecta en todas las regiones que se conservan. Por ejemplo, ignorando todos los demás efectos, la probabilidad de que un 12—mer permanezca fijo en todas las 29 especies es menor que 10-25. Así, las regiones que están casi perfectamente conservadas en múltiples especies se destacan por ser únicas y dignas de estudio adicional. Una de esas regiones se muestra en la Figura 4.6.
Causas del exceso de restricción
La pregunta es ¿qué presiones evolutivas hacen que ciertas regiones estén tan perfectamente conservadas? Las siguientes fueron todas mencionadas en clase como posibilidades:
- ¿Podría ser que exista una estructura especial de ADN que proteja esta zona de la mutación?
- ¿Hay alguna maquinaria especial para corregir errores que se asiente en este lugar?
- ¿Puede la célula utilizar el estado de metilación de las dos copias de ADN como mecanismo de corrección de errores? Este mecanismo se basaría en el hecho de que la nueva copia de ADN no está metilada, y por lo tanto la maquinaria de replicación del ADN podría verificar la nueva copia contra la copia vieja metilada.
- ¿Quizás la próxima generación no pueda sobrevivir si esta región está mutada?
Otra posible explicación es que se está produciendo selección para conservar codones específicos. Algunos codones son más eficientes que otros: por ejemplo, las proteínas de mayor abundancia que necesitan una traducción rápida pueden seleccionar codones que dan la tasa de traducción más eficiente, mientras que otras proteínas pueden seleccionar codones que dan una traducción menos eficiente.
Aún así, estas regiones parecen estar demasiado perfectamente conservadas para ser explicadas solo por el uso de codones. ¿Qué más puede explicar el exceso de restricción? Debe haber algún grado de precisión necesario a nivel de nucleótidos que impida que estas secuencias diverjan.
Podría ser que estemos viendo la misma región en dos especies que apenas han divergido recientemente o que existe un mecanismo genético específico que proteja esta zona. Sin embargo, es más probable que tanta conservación sea un signo de regiones codificantes de proteínas que codifican simultáneamente otros elementos funcionales. Por ejemplo, el gen HOXB5 muestra un obvio exceso de restricción, y hay evidencia de que el extremo 5' del ORF de HOXB5 codifica tanto proteína como una estructura secundaria de ARN.
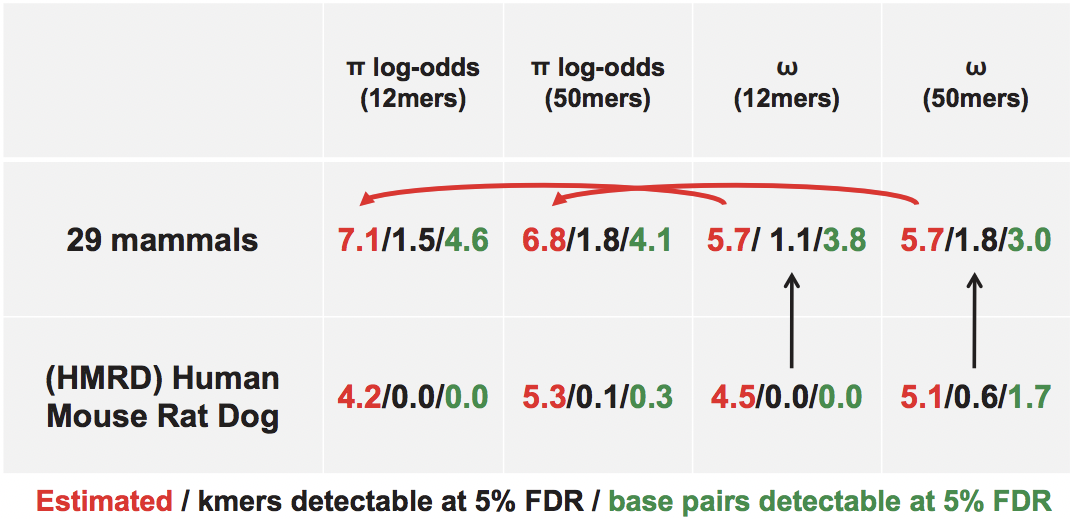
Figura 4.5: Al aumentar el número de mamíferos estudiados, vemos un aumento en los k-meros restringidos y pares de bases que son detectables.
Las regiones que codifican más de un tipo de elemento funcional están bajo presiones selectivas superpuestas. Podría haber presión en el espacio de codificación de proteínas para mantener la secuencia de aminoácidos correspondiente a esta región igual, combinada con la presión del espacio de ARN para mantener una secuencia de nucleótidos que preserva la estructura secundaria del ARN. Como resultado de estas dos presiones para mantener codones para los mismos aminoácidos y producir la misma estructura de ARN, es probable que la región muestre mucha menos tolerancia para cualquier patrón de sustitución sinónimo.
El proceso de estimación de restricción evolutiva a partir de datos de alineamiento genómico en múltiples especies sigue los siguientes pasos:
- Contar el número de operaciones de edición (es decir, el número de sustituciones y/o eliminaciones/inserciones)
- Estimar el número de mutaciones incluyendo retromutaciones
- Incorporar información sobre los elementos vecinales del elemento conservado mirando” ventanas de conservación”
- Estimar la probabilidad de un “estado oculto” restringido mediante el uso de Modelos Ocultos de Markov
- Utilice la filogenia para estimar la tasa de mutación del árbol (es decir, rechazar sustituciones que deberían ocurrir a lo largo del
árbol)
- Permitir que diferentes porciones del árbol tengan diferentes tasas de mutación
Modelado de restricción de exceso
Para estudiar mejor la región de restricción excesiva, desarrollamos modelos matemáticos para medir sistemáticamente la cantidad de conservación sinónima y no sinónima de diferentes regiones. Mediremos dos tasas: conservación de codones y nucleótidos.
Para representar el modelo nulo, podemos construir matrices de velocidad (4 × 4 en el caso de nucleótidos y 64 × 64 para el caso del codón) que dan las tasas de sustituciones entre codones o nucleótidos por unidad de tiempo. Estimamos las tasas en el modelo nulo observando una tonelada de datos y estimando las probabilidades de cada tipo de sustitución. Véase la Figura 4.18a en 4.5.2 para un ejemplo de una matriz nula para el caso del codón.
• λs: la tasa de sustituciones sinónimas

Figura 4.6: Muchas regiones genómicas, como HOXB5, muestran más conservación de la que esperaríamos en regiones codificantes conservadas normales. Entre las 29 especies en estudio, todas menos 7 de ellas tenían exactamente la misma secuencia de nucleótidos. Las áreas verdes son áreas que han sufrido mutaciones evolutivas.

Figura 4.7: Podemos modelar mutaciones usando matrices de velocidad, como se muestra aquí para las sustituciones de nucleótidos a la izquierda y las sustituciones de codones a la derecha. En cada matriz, la celda en la fila m y la enésima columna representa la probabilidad de que el símbolo m mute en el símbolo n. Cuanto más oscuro es el color, menos probable es la mutación.
Por ejemplo, si λs = 0.5, entonces la tasa de sustituciones sinónimas es la mitad de lo que se espera del modelo nulo en esa región. Luego podemos evaluar la significancia estadística de las estimaciones de tasa que obtenemos, y encontrar regiones donde la tasa de sustitución es mucho menor de lo esperado.
El uso de un modelo nulo aquí nos ayuda a dar cuenta de los sesgos en la cobertura de alineación de ciertos codones y también da cuenta de la posibilidad de degeneración de codones, en cuyo caso esperaríamos ver una tasa mucho mayor de sustituciones. Aprenderemos a combinar dichos modelos con métodos filogénicos cuando hablemos de árboles filogénicos y evolución más adelante en el curso.
La aplicación de este modelo muestra que las secuencias en los primeros codones traducidos, exones de casete (exones que están presentes en un transcrito de ARNm pero ausentes en una isoforma del transcrito) y, alternativamente, regiones empalmadas tienen tasas especialmente bajas de sustituciones sinónimos.
Exceso de restricción en el genoma humano
En esta sección, examinaremos el problema de determinar la proporción total del genoma humano bajo restricción excesiva. En particular, revisitaremos el trabajo de Lindblad—Toh et al. (2011), que compararon 29 genomas de mamíferos. Midieron los niveles de conservación a lo largo del genoma aplicando el proceso descrito en la sección anterior a 50—meros. Al considerar solo 50—meros que formaban parte de repeticiones ancestrales, es posible determinar un nivel de fondo de conservación. Podemos imaginar que las intensidades de conservación entre los 50 meros se distribuyen de acuerdo con una distribución de probabilidad oculta, como se ilustra en la Figura 4.8. En la figura, la curva de fondo representa la distribución de restricción en ausencia de mecanismos especiales para el exceso de restricción, según se determina al observar repeticiones ancestrales, mientras que la curva de señal (primer plano) representa la distribución real del genoma. La curva de señal tiene más conservación en general debido a los efectos purificadores de la selección natural.
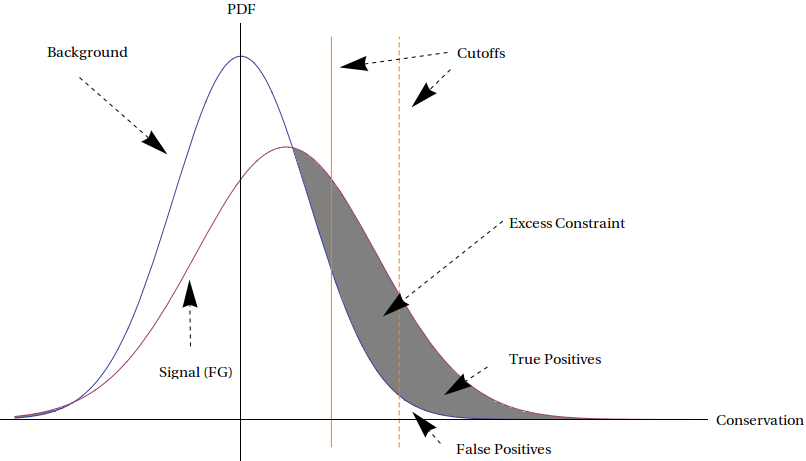
Figura 4.8: Medición del exceso de restricción genómica amplia. Consulte el texto adjunto para una explicación.
Es posible que deseemos investigar regiones específicas del genoma que están bajo restricciones excesivas estableciendo un nivel umbral de conservación y examinando las regiones que están más conservadas. En la ilustración, esto corresponde a considerar todos los 50—mers que caen a la derecha de una de las líneas anaranjadas. Vemos que si bien este método efectivamente nos da regiones bajo restricción excesiva, también nos da falsos positivos. Esto se debe a que incluso en ausencia de selección purificadora y otros efectos, ciertas regiones estarán fuertemente conservadas, simplemente por casualidad aleatoria. Establecer el umbral más alto, como usar la línea punteada naranja como nuestro umbral, reduce la proporción de falsos positivos (FP) a verdaderos positivos (TP), al tiempo que disminuye el número de verdaderos positivos detectados, negociando así mayor especificidad por menor sensibilidad.
No obstante, no toda esperanza está perdida. Es posible medir empíricamente las curvas de señal tanto de fondo (BG) como de primer plano (FG), como se describió anteriormente. Una vez hecho esto, el área de la región entre ellos, que está sombreada en gris en la Figura 4.8, se puede determinar por integración. Esta área representa la proporción del genoma que está bajo restricción excesiva. Debido a que las curvas se superponen, no podemos detectar todos los elementos conservados pero podemos estimar la cantidad total de restricción excesiva. Este número de restricción estimada resulta ser alrededor del 5% del genoma humano, dependiendo de qué tan grande se use una ventana. Es probable que todas esas regiones sean funcionales, pero dado que alrededor del 1.5% del genoma humano es codificante de proteínas, podemos inferir que el 3.5% restante consiste en elementos funcionales no codificantes, la mayoría de los cuales probablemente desempeñan funciones reguladoras.
Hemos visto que la restricción evolutiva sobre todo el genoma se puede estimar evaluando la restricción genómica contra una distribución de fondo. Lindblad-Toh et al. (2011) comparan la conservación del genoma en 29 mamíferos frente a un fondo calculado a partir de elementos de repetición ancestrales para encontrar regiones con restricción excesiva (Figura 4.9A y B). La anotación de bases evolutivamente restringidas revela que la mayoría de las regiones descubiertas son intergénicas e intrónicas y demuestra que pasar de cuatro (HMRD) a 29 genomas de mamíferos aumenta el poder de este análisis principalmente en regiones no codificantes (Figura 4.9C). Las regiones más restringidas en el genoma son regiones codificantes (Figura 4.9D).
Como se muestra en la Figura 4.9, el aumento de HMRD a una alineación de 29 genomas mejora enormemente la potencia
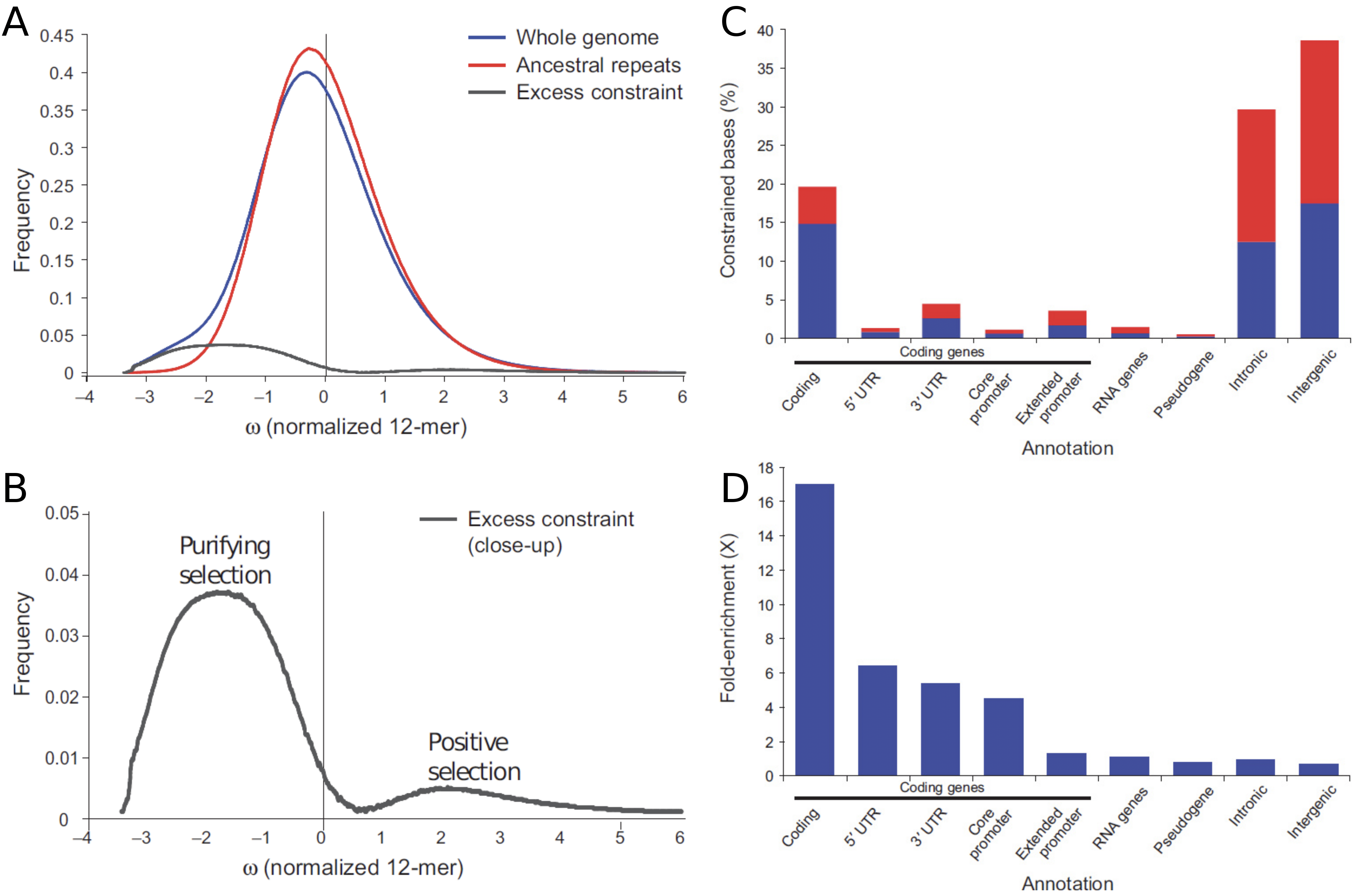
Figura 4.9: Detectando elementos funcionales a partir de su firma evolutiva. A Distribución de restricción para todo el genoma frente a repeticiones ancestrales (fondo). B Diferencia entre el genoma completo y la restricción de fondo. C Descubrimiento de elementos funcionales a partir del exceso de restricción. Los elementos novedosos se muestran en rojo. D Enriquecimiento de elementos para regiones de restricción excesiva.
de este análisis. Sin embargo, si bien la cantidad de elementos intergénicos detectados aumentó significativamente, la detección aún está limitada por el hecho de que los elementos no funcionales tienen una profundidad de cobertura de especies mucho menor en múltiples alineamientos que las regiones funcionales (Figura 4.10). Por ejemplo, las repeticiones ancestrales (AR, μ = 11.4) tienen una profundidad de cobertura promedio mucho menor que los exones (μ = 20.9). Por un lado, esto muestra evidencia de selección contra inserciones y deleciones en elementos funcionales, las cuales no son examinadas en el análisis de restricción base. Por otro lado, también complica el análisis de la restricción evolutiva, ya que dicho trabajo debe entonces manejar una cobertura variable a lo largo del genoma.
Ejemplos de Restricción Exceso
Se han encontrado ejemplos de restricción excesiva en los siguientes casos:
- La mayoría de los genes Hox muestran regiones de restricción superpuestas. En particular, como se mencionó anteriormente los primeros 50 aminoácidos de HOXB5 están casi completamente conservados. Además, HOXA2 muestra módulos regulatorios superpuestos. Estos dos loci codifican potenciadores del desarrollo, proporcionando un mecanismo para la expresión específica del tejido.
- ADAR: el principal regulador de la edición de ARNm, tiene una variante de empalme donde se encontró una baja tasa de sustitución sinónima a una resolución de 9 codones.
- BRCA1: Hurst y Pal (2001) encontraron una baja tasa de sustituciones sinónimas en ciertas regiones del BRCA1, el principal gen involucrado en el cáncer de mama. Ellos plantearon la hipótesis de que la selección purificadora se produce- anillo en estas regiones. (Esta afirmación fue refutada por Schmid y Yang (2008) quienes afirman que este fenómeno es el artefacto de un análisis de ventana deslizante).
- THRA/NR1D1: estos genes, también involucrados en el cáncer de mama, forman parte de una región codificadora dual que codifica para ambos genes y está altamente conservada.
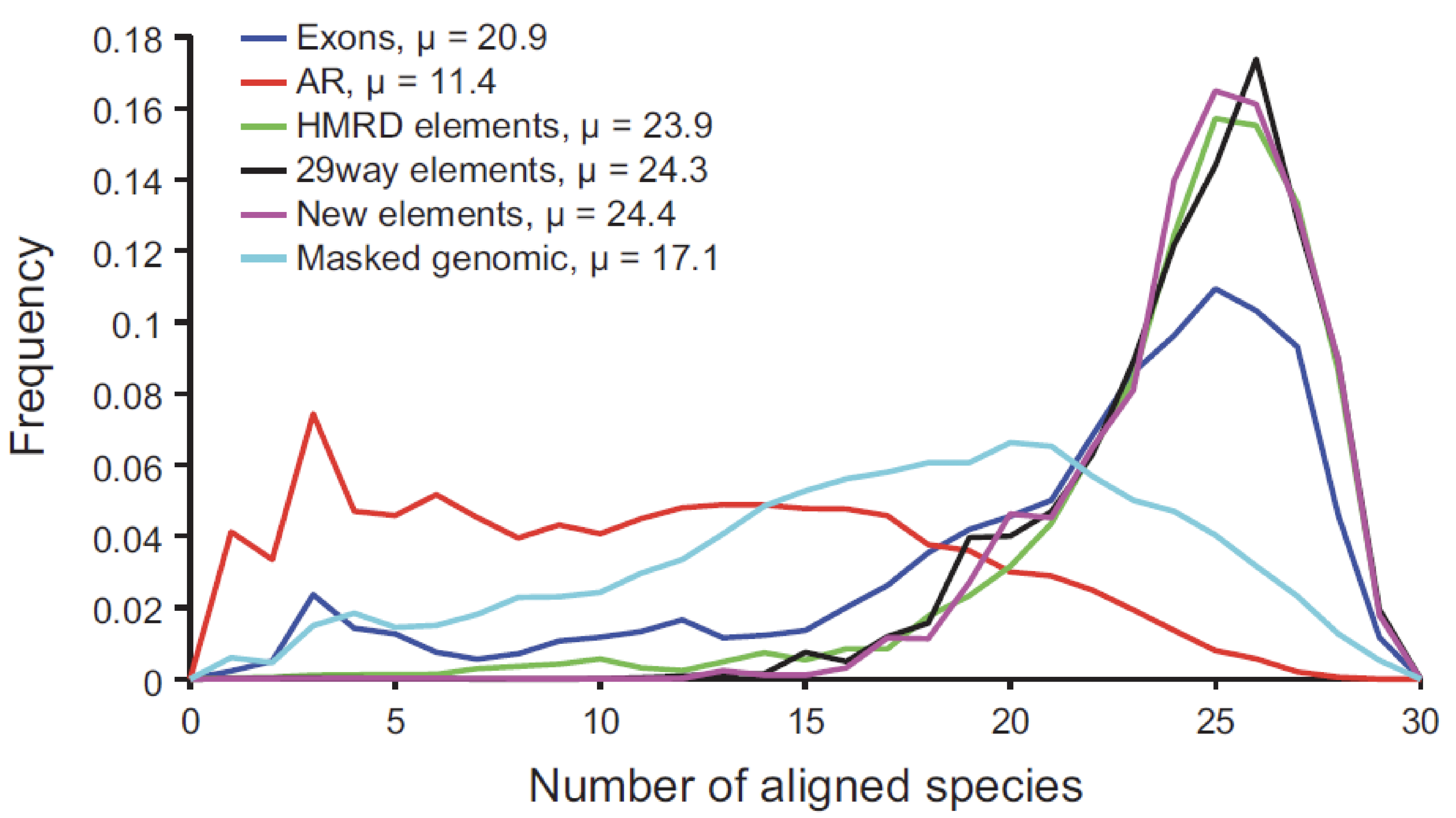
Figura 4.10: Profundidad de cobertura a través de diferentes conjuntos de elementos.
- SEPHS2: tiene una horquilla involucrada en la recodificación de selenocisteína. Debido a que esta región debe seleccionar codones tanto para conservar la secuencia de aminoácidos de la proteína como los nucleótidos para mantener la misma estructura secundaria de ARN, muestra un exceso de restricción.
Medición de restricción en nucleótidos individuales
Al medir la restricción evolutiva en nucleótidos individuales en lugar de bloques de la secuencia, podemos encontrar sitios de unión a factores de transcripción individuales, sesgo específico de posición dentro de instancias de motivos y revelar consenso de motivo entre la mayoría de las especies. Específicamente, podemos detectar SNP que interrumpen los motivos reguladores conservados y determinar el nivel de evolución observando cada nucleótido en el gen. Al observar los nucleótidos individualmente, podemos encontrar SNP que son importantes en la función de una secuencia específica.
2 www.nature.com/nature/journal... ture10530.html