
4.5: Firmas de codificación de proteínas
- Page ID
- 54784

En la diapositiva 12, vemos tres ejemplos de conservación: una secuencia intrónica con mala conservación, una región codificante con alta conservación y una región no codificante con alta conservación, lo que significa que probablemente sea un elemento funcional. Como vimos al inicio de esta sección, la característica importante de las regiones codificadoras de proteínas para recordar es que los codones (triples de nucleótidos) codifican los aminoácidos, que componen las proteínas. Esto da como resultado la firma evolutiva de las regiones codificadoras de proteínas, como se muestra en la diapositiva 13: (i) conservación del marco de lectura y (ii) patrones de sustitución de codones. La intuición para esta firma es relativamente sencilla.
Figura 4.14: Algunas mutaciones puntuales a la secuencia de ADN no cambian la traducción de proteínas
En primer lugar, la conservación del marco de lectura tiene sentido, ya que una inserción o deleción de uno o dos nucleótidos “desplazará” la forma en que se leen todos los siguientes codones. Sin embargo, si ocurre una inserción o eliminación en un múltiplo de 3, los otros codones seguirán siendo leídos de la misma manera, por lo que este es un cambio menos significativo. En segundo lugar, tiene sentido que algunas mutaciones sean menos dañinas que otras, ya que diferentes tripletes pueden codificar para los mismos aminoácidos (una sustitución conservadora, como se desprende de la matriz de abajo), e incluso las mutaciones que dan como resultado un aminoácido diferente pueden ser evolutivamente neutras si se producen las sustituciones con aminoácidos similares en un dominio de la proteína donde no se requieren las propiedades exactas de los aminoácidos. Estos patrones distintivos nos permiten “colorear” el genoma y ver claramente dónde están los exones, como se muestra en la Figura 4.15.
Al usar estos patrones para distinguir las firmas evolutivas, tenemos que asegurarnos de considerar las siguientes ideas:
Figura 4.15: Al colorear los tipos de inserciones/deleciones/sustituciones que ocurren en una secuencia, podemos ver patrones o firmas evolutivas que distinguen una región conservada codificante de proteína de una región conservada que no codifica proteínas.
- Cuantificar la distinción de las 64 2 posibles sustituciones de codones considerando regiones sinónimas (frecuentes en secuencias codificantes de proteínas) y sin sentido (más frecuentes en secuencias no codificantes que codificantes).
- Modelar la relación filogenética entre las especies: múltiples sustituciones aparentes pueden ser ex- plaqueadas por un evento evolutivo.
- Tolerar la incertidumbre en la entrada como secuencias ancestrales desconocidas y brechas en alineación (datos faltantes).
- Informar la certeza o incertidumbre del resultado: cuantificar la confianza de que una alineación dada es codificadora de proteínas utilizando diversas unidades como valor p, bits, decibanos... etc.
Lectura: conservación de marco (RFC)
Ahora que conocemos este patrón de conservación en genes codificadores de proteínas, podemos desarrollar métodos para determinar si un gen es codificante de proteínas o si no lo es.
Al puntuar la presión para permanecer en el mismo marco de lectura, podemos cuantificar la probabilidad de que una región sea codificadora de proteínas o no. Como se muestra en la diapositiva 20, podemos hacer esto teniendo una secuencia diana (Scer, el genoma de S. cerevisiae), y luego alinear una secuencia de selección (Spar, S. paradoxus) con ella y calculando qué proporción del tiempo la secuencia seleccionada coincide con el marco de lectura de la secuencia diana.
Como no sabemos dónde comienza el marco de lectura en la secuencia seleccionada, alineamos tres veces para probar todas las compensaciones posibles:
(Sparf1, Sparf2, Sparf3)
A partir de estos, elegimos el alineamiento donde la secuencia seleccionada suele estar sincronizada con la secuencia diana. Por ejemplo, podemos comenzar a numerar los nucleótidos “1, 2, 3... etc.” hasta llegar a una brecha que no numeramos. O podemos comenzar a numerar los nucleótidos “2, 3, 1... etc.” donde cada triplete de “1,2,3” representa un codón.
Finalmente, para el mejor alineamiento, calculamos el porcentaje de nucleótidos que están fuera de marco —si está por encima de un límite, esta especie seleccionada “vota” que esta región es una región codificadora de proteínas, y si es baja, esta especie “vota” que se trata de una región intergénica. Se contabilizan los “votos” de todas las especies para sumarse al puntaje RFC.
Figura 4.16: Dos alineamientos que muestran diferencias de patrones de conservación entre secuencias génicas e intergénicas. Los cuadros rojos representan huecos que desplazan el marco de codificación, y los cuadros grises son huecos que no cambian de cuadro (en múltiplos de tres). Las regiones verdes se conservan y las amarillas están mutadas. Obsérvese el patrón de “coincidencia, coincidencia, falta de coincidencia” en la secuencia codificante de proteínas que indica mutaciones sinónimas.
Este método no es robusto al error de secuenciación. Podemos compensar estos errores usando una ventana de escaneo más pequeña y observando la conservación del marco de lectura local.
Se demostró que el método tiene 99.9% de especificidad y 99% de sensibilidad cuando se aplica al genoma de levadura. Cuando se aplicó a 2000 ORF hipotéticos (marcos de lectura abiertos, o genes propuestos) 3 en levaduras, rechazó 500 de estos supuestos genes codificantes de proteínas por no ser codificantes de proteínas.
De igual manera, 4000 genes hipotéticos en el genoma humano fueron rechazados por este método. Este modelo creó una hipótesis específica (que era poco probable que estas secuencias de ADN codificaran proteínas) que posteriormente se ha apoyado con la confirmación experimental de que las regiones no codifican proteínas in vivo. 4
Esto representa un importante paso adelante para la anotación del genoma, ya que anteriormente era difícil concluir que una secuencia de ADN no codificaba simplemente por falta de evidencia. Al reducir el enfoque y crear una nueva hipótesis nula (que el gen en cuestión parece ser un gen no codificante) se hizo mucho más fácil no solo aceptar genes codificantes, sino rechazar los genes no codificantes con soporte computacional. Durante la discusión sobre la conservación del marco de lectura en clase, identificamos una idea emocionante para un proyecto final que sería buscar el nacimiento de nuevas proteínas funcionales resultantes de mutaciones de cambio de marco.
Frecuencias de sustitución de codones (CSF)
La segunda firma de las regiones codificadoras de proteínas, las frecuencias de sustitución de codones, actúa sobre múltiples niveles de conservación. Para explorar estas frecuencias, es útil recordar que la evolución de codones puede ser modelada
Figura 4.17: Los cuadros rojos representan huecos de desplazamiento de marco, y los huecos en múltiplos de tres son incoloros. Las regiones conservadas y mutadas son verdes y amarillas, respectivamente.
por distribuciones de probabilidad condicional (CPD) — la probabilidad de que un descendiente tenga un codón b donde un antepasado tenía el codón a una cantidad de tiempo t ago.
El evento más conservador es el mantenimiento exacto del codón. Una mutación que codifica para el mismo aminoácido puede ser conservadora pero no totalmente sinónimo, debido a los sesgos de uso de codones específicos de especies. Incluso las mutaciones que alteran la identidad del aminoácido podrían ser conservadoras si codifican aminoácidos con propiedades bioquímicas similares.
Utilizamos un DPC para capturar el efecto neto de todas estas consideraciones. Para calcular estos CPD, necesitamos una matriz de “tasa”, Q, que mida el tipo de cambio para una unidad de tiempo; es decir, indica con qué frecuencia el codón a en la especie 1 se sustituye por el codón b en la especie 2, por una longitud de rama unitaria. Entonces, usando e Qt, podemos estimar la frecuencia de sustitución en el tiempo t.
Cuando el CPD se considera en conjunto con la topología de una gráfica de red que representa el árbol evolutivo, tiene aproximadamente (2L − 2) · 64 2 parámetros, donde L es el número de hojas en el árbol (especies en la filogenia evolutiva). Este número de parámetros se deriva del número de entradas en Q y el número de longitudes de rama independientes, t. Las estimaciones de estos parámetros pueden ser determinadas por MLE a partir de datos de entrenamiento.
El CPD se define en términos de e Qt de la siguiente manera:
\ [\ begin {ecuación}
\ operatorname {Pr} (\ text {child} =a\ mid\ text {parent} =b; t) =\ left [e^ {Q t}\ right] _ {a, b}
\ end {ecuación}\]
La intuición, es que a medida que aumenta el tiempo, aumenta la probabilidad de sustituciones, mientras que en el tiempo “inicial” (t = 0), e Qt es la matriz de identidad, ya que se garantiza que cada codón sea él mismo. Pero, ¿cómo obtenemos la matriz de tarifas?
- Q se “aprende” de las secuencias, mediante el uso de Expectación-Maximización, por ejemplo. Muchas secuencias codificadoras de proteínas conocidas se utilizan como datos de entrenamiento (o regiones no codificantes al generar ese modelo).
- Dados los parámetros del modelo, podemos usar el algoritmo de Felsenstein [1] para calcular la probabilidad de cualquier alineación, teniendo en cuenta la filogenia, dado el modelo de sustitución (el paso E).
\ [\ begin {ecuación}
\ text {Probabilidad} (\ boldsymbol {Q}) =\ nombreoperador {Pr} (\ text {Datos de entrenamiento;}\ símbolo en negrilla {Q}, t)
\ end {ecuación}\]
- Entonces, dadas las alineaciones y la filogenia, podemos elegir los parámetros (la matriz de tasa: Q, y longitudes de rama: t) que maximicen la probabilidad de esas alineaciones en el paso M; por ejemplo, para estimar Q, podemos contar el número de veces que un codón es sustituido por otro en el alineamiento. El espacio de argumento consiste en miles de posibilidades para Q y t. Este espacio está representado por Q.\ (\ begin {ecuación}
\ hat {Q}
\ end {ecuación}\) es el parámetro que maximiza la probabilidad:
\ [\ begin {ecuación}
\ hat {Q} =\ nombreoperador {argmax} _ {\ boldsymbol {Q}} (\ text {Probabilidad} (Q))
\ end {ecuación}\]
Otras estrategias de maximización incluyen: maximización de expectativas, ascenso de gradiente, recocido simulado, descomposición espectral. La longitud de la rama, t, se puede optimizar usando el mismo método simultáneamente.
FAQ
P: ¿Cómo contribuye la longitud de la sucursal a determinar la matriz de tasas?
R: Las longitudes de las ramas especifican cuánto “tiempo” pasó entre dos nodos cualesquiera. La matriz de velocidad describe las frecuencias relativas de sustituciones de codones por unidad de longitud de rama.
Con dos matrices de tasas estimadas, las probabilidades calculadas de cualquier alineación dada son diferentes para cada matriz. Ahora, podemos comparar la razón de verosimilitud\(\frac{\operatorname{Pr}\left(\text {Leaves} ; Q_{c}, t\right)}{\operatorname{Pr}\left(\text {Leaves} ; Q_{N}, t\right)}\),, que el alineamiento vino de una región codificante de proteínas en lugar de provenir de una región que no codifica proteínas.
© fuente desconocida. Todos los derechos reservados. Este contenido está excluido de nuestra licencia Creative Commons. Para obtener más información, consulte http://ocw.mit.edu/help/faq-fair-use/.
Figura 4.18: Matrices de tasas para los modelos nulos y alternos. Un color más claro significa que la sustitución es más probable.
Ahora que sabemos cómo obtener nuestro modelo, observamos que, dado el patrón específico de frecuencias de sustitución de codones para la codificación de proteínas, queremos dos modelos para que podamos distinguir entre regiones codificantes y no codificantes. Las figuras 4.18a y 4.18b muestran matrices de tasas para regiones intergénicas y génicas, respectivamente. Una serie de características sobresalientes se presentan en la matriz de sustitución de codones (CSM) para genes. Obsérvese que se ha eliminado el elemento diagonal principal, ya que la frecuencia de un triplete que se intercambia por sí mismo obviamente será mucho mayor que cualquier otro intercambio. Sin embargo,
- inmediatamente es obvio que existe un fuerte elemento diagonal en las regiones codificantes de proteínas.
- También observamos ciertos elementos diagonales de alta puntuación en el CSM codificante: estas son sustituciones que tienen una función más cercana que en secuencia, como codones degenerados de 6 veces o aminoácidos muy similares.
- También observamos franjas verticales oscuras, que indican que estas sustituciones son especialmente improbables. Estas columnas corresponden a codones de parada, ya que las sustituciones a este triplete alterarían significativamente la función de la proteína, y por lo tanto se seleccionan fuertemente contra.
Por otro lado, en la matriz para regiones intergénicas, los tipos de cambio son más uniformes. En estas regiones, lo que importa es la proximidad mutacional, es decir, la distancia de edición o el número de cambios de una secuencia a otra. Las regiones genéticas están dictadas por la proximidad selectiva, o la similitud en la secuencia de aminoácidos de la proteína resultante del gen.
Ahora que tenemos las dos matrices de tasas para las dos regiones, podemos calcular las probabilidades de que cada matriz genere los genomas de las dos especies. Esto se puede hacer usando el algoritmo de Felsenstein, y sumando la “puntuación” para cada par de codones correspondientes en las dos especies. Finalmente, podemos calcular la razón de verosimilitud de que el alineamiento vino de una región codificante a una región no codificante dividiendo las dos puntuaciones, esto demuestra nuestra confianza en nuestra anotación de la secuencia. Si la relación es mayor que 1, podemos adivinar que es una región codificante, y si es menor que 1, entonces es una región no codificante. Por ejemplo, en la Figura 4.16, tenemos mucha confianza en las respectivas clasificaciones de cada región.
Cabe señalar, sin embargo, que aunque la “coloración” de las secuencias confirma nuestras clasificaciones, las razones de verosimilitud se calculan independientemente de la 'coloración', que utiliza nuestro conocimiento de sustituciones sinónimas o conservadoras. Esto implica además que este método infiere automáticamente el código genético a partir del patrón de sustituciones que se produce, simplemente observando las sustituciones de alta puntuación. En especies con un código genético diferente, los patrones de intercambio de codones serán diferentes; por ejemplo, en la albúmina de Candida, el CTG codifica serina (polar) en lugar de leucina (hidrófoba), y esto se puede deducir de los CSM. Sin embargo, el método no requiere ningún conocimiento de esto; en cambio, podemos deducir esto a posteriori del CSM.
En resumen, podemos distinguir entre regiones no codificantes y codificantes del genoma en función de sus firmas evolutivas, mediante la creación de dos matrices separadas de 64 por 64 tasas: una que mide la tasa de sustituciones de codones en regiones codificantes y la otra en regiones no codificantes. La matriz de tasas da la tasa de cambio de codones o nucleótidos a lo largo de una unidad de tiempo.
Se utilizaron las dos matrices para calcular dos probabilidades para cualquier alineación dada: la probabilidad de que proviniera de una región codificante y la probabilidad de que proviniera de una región no codificante. Tomando la razón de verosimilitud de estas dos probabilidades da una medida de confianza de que el alineamiento es proteína, codificando como estrato demoníaco en la Figura 4.19. Mediante este método podemos seleccionar regiones del genoma que evolucionan de acuerdo con la firma codificante de la proteína.
puntuaciones — esto demuestra nuestra confianza en nuestra anotación de la secuencia. Si la relación es mayor que 1, podemos adivinar que es una región codificante, y si es menor que 1, entonces es una región no codificante. Por ejemplo, en la Figura 4.16, tenemos mucha confianza en las respectivas clasificaciones de cada región.
Cabe señalar, sin embargo, que aunque la “coloración” de las secuencias confirma nuestras clasificaciones, las razones de verosimilitud se calculan independientemente de la 'coloración', que utiliza nuestro conocimiento de sustituciones sinónimas o conservadoras. Esto implica además que este método infiere automáticamente el código genético a partir del patrón de sustituciones que se produce, simplemente observando las sustituciones de alta puntuación. En especies con un código genético diferente, los patrones de intercambio de codones serán diferentes; por ejemplo, en la albúmina de Candida, el CTG codifica serina (polar) en lugar de leucina (hidrófoba), y esto se puede deducir de los CSM. Sin embargo, el método no requiere ningún conocimiento de esto; en cambio, podemos deducir esto a posteriori del CSM.
En resumen, podemos distinguir entre regiones no codificantes y codificantes del genoma en función de sus firmas evolutivas, mediante la creación de dos matrices separadas de 64 por 64 tasas: una que mide la tasa de sustituciones de codones en regiones codificantes y la otra en regiones no codificantes. La matriz de tasas da la tasa de cambio de codones o nucleótidos a lo largo de una unidad de tiempo.
Se utilizaron las dos matrices para calcular dos probabilidades para cualquier alineación dada: la probabilidad de que proviniera de una región codificante y la probabilidad de que proviniera de una región no codificante. Tomando la razón de verosimilitud de estas dos probabilidades da una medida de confianza de que el alineamiento es proteína, codificando como estrato demoníaco en la Figura 4.19. Mediante este método podemos seleccionar regiones del genoma que evolucionan de acuerdo con la firma codificante de la proteína.
Figura 4.19: Como podemos ver en la figura que la razón de verosimilitud es positiva para secuencias que probablemente sean codificantes de proteínas y negativas para secuencias que probablemente no sean codificantes de proteínas.
Veremos más adelante cómo combinar este enfoque de cociente de verosimilitud con métodos filogenéticos para encontrar patrones evolutivos de regiones codificantes de proteínas.
Sin embargo, este método solo nos permite encontrar regiones que se seleccionan a nivel traslacional. El punto clave es que aquí estamos midiendo solo para la selección de codificación de proteínas. Veremos hoy cómo podemos buscar otros elementos funcionales conservados que exhiban sus propias firmas únicas.
Clasificación de las secuencias del genoma de Drosophila
Hemos visto que el uso de estas métricas de RFC y CSF nos permite clasificar exones e intrones con especificidad y sensibilidad extremadamente altas. Los clasificadores que utilizan estas medidas para clasificar secuencias se pueden implementar usando un campo aleatorio condicional (SMCRF) HMM o Semi-Markov. Los CRF permiten la integración de diversas características que no necesariamente tienen una naturaleza probabilística, mientras que los HMM requieren que modelaremos todo como probabilidades de transición y emisión. Los CRF serán discutidos en una próxima conferencia. Uno podría preguntarse por qué es necesario implementar estos métodos más complejos, cuando el método más simple de verificar la conservación del marco de lectura funcionó bien. La razón es que en regiones muy cortas, las inserciones y deleciones serán muy poco frecuentes, incluso por casualidad, por lo que no habrá suficiente señal para hacer la distinción entre regiones codificantes de proteínas y no codificantes de proteínas. En la siguiente figura, vemos una secuencia de ADN a lo largo del eje x, con las filas que representan un gen anotado, cantidad de conservación, cantidad de proteína que codifica la firma evolutiva y el resultado de la decodificación de Viterbi usando el SMCRF, respectivamente.
Figura 4.20: Las firmas evolutivas pueden predecir nuevos genes y exones. La estrella denota un nuevo exón, el cual se predijo mediante las tres pruebas de genómica comparativa, y posteriormente se verificó mediante secuenciación de ADNc.
Este es un ejemplo de cómo la utilización de la firma codificante de proteínas para clasificar regiones ha demostrado ser muy exitosa. La identificación de regiones que se habían pensado que eran genes pero que no tenían alto contenido proteico, las firmas codificantes nos permitieron rechazar fuertemente 414 genes en el genoma de la mosca previamente clasificados como CGID, solo genes, lo que llevó a los curadores de FlyBase a eliminar 222 de ellos y marcar otros 73 como inciertos. Además, también hubo falsos negativos definitivos, ya que existía evidencia funcional para los genes bajo examen. Finalmente, en los datos, también vemos regiones con ambas conversaciones, así como una gran firma codificante de proteínas, pero no se habían marcado previamente como partes de genes, como en la Figura 4.20. Algunos de estos han sido probados experimentalmente y se ha demostrado que son partes de nuevos genes o extensiones de genes existentes. Esto subraya la utilidad de la biología computacional para apalancar y dirigir el trabajo experimental.
Codones de parada con fugas
Los codones de parada (TAA, TAG, TGA en ADN y UAG, UAA, UGA en ARN) suelen señalar el final de un gen. Reflejan claramente la terminación de la traducción cuando se encuentran en el ARNm y liberan la cadena de aminoácidos del ribosoma. Sin embargo, en algunos casos inusuales, la traducción se observa más allá del primer codón de parada. En casos de lectura única, hay un codón de terminación que se encuentra dentro de una región con una proteína clara que codifica la firma seguida de un segundo codón de parada a corta distancia. Un ejemplo de esto en el genoma humano se da en la Figura 4.21. Esto sugiere que la traducción continúa a través del primer codón de parada. También se han observado casos de doble lectura, donde dos codones de parada se encuentran dentro de una región codificante de proteínas. En estos casos de supresión de codones de terminación, se encuentra que el codón de parada está altamente conservado, lo que sugiere que estos codones de parada omitidos juegan un papel biológico importante.
La lectura traslacional se conserva en ambas moscas, que tienen 350 proteínas identificadas que exhiben lectura de codones de parada, y en humanos, que tienen 4 instancias identificadas de tales proteínas. Se observan principalmente en proteínas neuronales en cerebros adultos y proteínas expresadas en el cerebro en Drosophila.
El gen kelch exhibe otro ejemplo de supresión de codones de parada en el trabajo. El gen codifica dos ORF con un único codón de parada UGA entre ellos. De esta secuencia se traducen dos proteínas, una del primer ORF y otra de la secuencia completa. La proporción de las dos proteínas está regulada de una manera específica del tejido. En el caso del gen kelch, una mutación del codón de parada de UGA a UAA da como resultado una pérdida de función, lo que sugiere que la supresión del ARNt es el mecanismo detrás de la supresión del codón de parada.
Figura 4.21: Neurotransmisor OPRL1: uno de los cuatro nuevos candidatos de lectura traslacional en el genoma humano. Nótese que la región después del primer codón de parada exhibe una firma evolutiva similar a la de la región codificante antes del codón de terminación, lo que indica que el codón de terminación está “suprimido”.
Un ejemplo adicional de supresión de codones de parada es Caki, una proteína activa en la regulación de la liberación de neurotransmisores en Drosophila. Los marcos de lectura abiertos (ORF) son secuencias de ADN que contienen un codón de inicio y terminación. En Caki, leer el gen en el primer marco de lectura (Frame 0) da como resultado significativamente más ORF que leer en Frame 1 o Frame 2 (un exceso de ORF 440). En la Figura 4.22 se enumeran doce posibles interpretaciones para el exceso de ORF. Sin embargo, debido a que el exceso se observa solo en el Cuadro 0, solo son probables las primeras 4 interpretaciones:
- Lectura de codones de parada: el codón de parada se suprime cuando el ribosoma extrae ARNt que se empareja incorrectamente con el codón de parada.
- Tonterías recientes: Quizás alguna mutación sin sentido reciente está causando la lectura de codones de parada.
- A a I edición: A diferencia de lo que pensábamos anteriormente, el ARN todavía se puede editar después de la transcripción. En algunos casos la base A se cambia a una I, que puede leerse como una G. Esto podría cambiar un codón de parada de TGA a un TGG, que codifica un aminoácido. Sin embargo, este fenómeno sólo se encuentra en un par de casos.
- Selenocisteína, el “21º aminoácido”: A veces, cuando el codón TGA es leído por un cierto bucle que conduce a un pliegue específico del ARN, se puede decodificar como selenocisteína. Sin embargo, esto solo ocurre en cuatro proteínas de mosca, por lo que no se puede explicar toda la supresión de codones de parada.
Entre estos cuatro, tres de ellos (tonterías recientes, edición A a I, y selenocisteína) representan sólo 17 de los casos. De ahí que parezca que la lectura completa debe ser responsable de la mayoría, si no de todos, de los casos restantes. Además, se observa el uso de codones de parada sesgados, por lo que se descartan otros procesos como el corte y empalme alternativo (donde los exones de ARN después de la transcripción se reconectan de múltiples maneras que conducen a múltiples proteínas) o ORF independientes.
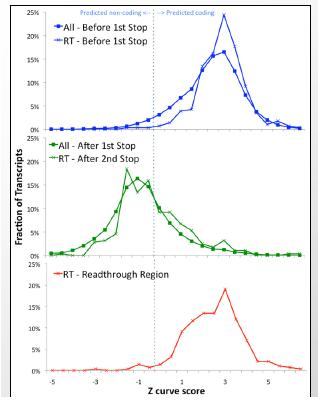
Las regiones de lectura pueden determinarse en una sola especie en función de su patrón de uso de codones. La curva Z, como se muestra en la Figura 4.23, mide los patrones de uso de codones en una región de ADN. A partir de la figura, se puede observar que la región de lectura coincide con la distribución antes del codón de parada regular. Sin embargo, después de la segunda parada, la región coincide con las regiones encontradas después de las paradas regulares.
Otra sugerencia ofrecida en clase fue la posibilidad de deslizamiento del ribosoma, donde el ribosoma salta algunas bases durante la traducción. Esto podría hacer que el ribosoma se salte más allá de un codón de parada. Este evento ocurre en genomas bacterianos y virales, los cuales tienen una mayor presión para mantener sus genomas pequeños, y por lo tanto pueden usar esta técnica de deslizamiento para leer una sola transcripción en cada marco de lectura diferente. Sin embargo, los humanos y las moscas no están bajo una presión tan extrema para mantener sus genomas pequeños. Adicionalmente, se demostró anteriormente que el exceso que observamos más allá del codón de parada es específico del marco 0, lo que sugiere que el deslizamiento del ribosoma no es responsable.
Las células son estocásticas en general y la mayoría de los procesos toleran errores a bajas frecuencias. El sistema no es perfecto y ocurren fugas de codones de parada. Sin embargo, la siguiente evidencia sugiere que la lectura de codones de parada no es aleatoria sino que está sujeta a control regulador:
- La conservación perfecta de los codones de parada de lectura se observa en 93% de los casos, lo que es muy superior al 24% encontrado en el fondo.
- Se observa un aumento de la conservación aguas arriba del codón de terminación de lectura.
Se observa sesgo de codón de parada. El TGAC es la secuencia más frecuente que se encuentra en el codón de parada en la lectura y la menos frecuente en los codones de terminación normales. Se sabe que es un codón de parada “permeable”. El TAAA se encuentra casi universalmente solo en instancias no leídas.
• Números inusualmente altos de repeticiones de GCA observados a través de codones de parada de lectura.
• El aumento de la estructura secundaria del ARN se observa después de la transcripción, lo que sugiere horquillas conservadas evolutivamente.
3 Kelis M, Patterson N, Endrizzi M, Birren B, Lander E. S. 2003. Secuenciación y comparación de especies de levaduras para identificar genes y elementos reguladores. Ciencia. 423:241—254.
4 Abrazadera M et al. 2007. Distinguir genes codificantes y no codificantes de proteínas en el genoma humano. PNAS. 104:19428—19433.