
8.3: Regulación de Pruebas Clínicas
- Page ID
- 52948

Diagnóstico in vitro (IVDs)
Los diagnósticos in vitro son pruebas que se realizan en muestras como sangre o tejido que han sido tomadas del cuerpo humano. Los diagnósticos in vitro pueden detectar enfermedades u otras afecciones y pueden usarse para monitorear la salud general de una persona para ayudar a curar, tratar o prevenir enfermedades. El diagnóstico in vitro también se puede utilizar en medicina de precisión para identificar a los pacientes que probablemente se beneficien de tratamientos o terapias específicas. Estos diagnósticos in vitro pueden incluir pruebas de secuenciación de próxima generación, que escanean el ADN de una persona para detectar variaciones genómicas. Algunas pruebas se utilizan en laboratorio u otros entornos profesionales de la salud y otras pruebas son para que los consumidores las usen en casa.
Supervisión del IVD
Bajo el acuerdo intercentro, tanto CDRH como CBER supervisan IVD. La Oficina de Investigación y Revisión de Sangre (OBRR) dentro del CBER gestiona la revisión previa al mercado y la vigilancia poscomercialización para IVDs asignados a CBER, mientras que la Oficina de Evaluación y Seguridad de Dispositivos de Diagnóstico In Vitro (OIVD) dentro de CDRH administra la revisión previa a la comercialización y la vigilancia poscomercialización para IVD asignado a CDRH. OIVD también es responsable de las exenciones de CLIA (ver más abajo). Los fabricantes solicitan la determinación de CLIA durante el proceso de revisión previa a la comercialización.
Clasificación IVD
Los IVD pueden clasificarse I, II o III dependiendo de su aplicación, diagnóstico, monitoreo, población de pacientes, tipo de espécimen y la consecuencia de un resultado falso de la prueba. Por ejemplo, si la prueba falsa da como resultado la amputación de una pierna por sospecha de cáncer, esta se clasificaría como un IVD Clase III. Aproximadamente el 8% de los IVD en el mercado son de Clase III. Esta clasificación determina la vía regulatoria para el dispositivo. www.fda.gov/medical-devices/ivd-regulatory-assistance/overview-ivdregulation
Hay 11 tipos de IVD listados en el sitio web de la FDA. Haga clic aquí y explórelos (panel en el lado izquierdo): https://www.fda.gov/medical-devices/products-and-medical-procedures/vitro-diagnostics. Aquí hay cinco interesantes y relevantes:
- Diagnóstico complementario: Un diagnóstico complementario es un dispositivo médico, a menudo un dispositivo in vitro, que proporciona información que es esencial para el uso seguro y efectivo de un medicamento o producto biológico correspondiente. La prueba ayuda a un profesional de la salud a determinar si los beneficios de un producto terapéutico para los pacientes superarán los efectos secundarios o riesgos potencialmente graves. Si la prueba diagnóstica es inexacta, entonces la decisión de tratamiento basada en esa prueba puede no ser óptima. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm407297.htm Los diagnósticos
complementarios pueden:- Identificar a los pacientes que tienen más probabilidades de beneficiarse de un producto terapéutico;
- identificar a los pacientes que probablemente tengan un mayor riesgo de sufrir efectos secundarios graves como resultado del tratamiento con un producto terapéutico, o
- monitorear la respuesta al tratamiento con un producto terapéutico particular con el propósito de ajustar el tratamiento para lograr una mayor seguridad o efectividad” (FDA.gov)
- Direct-to-Consumer: Los diagnósticos in vitro (IVD) que se comercializan directamente a los consumidores sin la participación de un proveedor de atención médica se denominan pruebas directas al consumidor (también denominadas DTC). Estas pruebas generalmente solicitan que el consumidor recoja una muestra, como saliva u orina, y la envíe a la empresa para su prueba y análisis. www.fda.gov/medical-devices/vitrodiagnostics/direct-consumer-tests Los diagnósticos
complementarios pueden:- Las pruebas directas al consumidor están expandiendo el número de personas que pueden hacerse pruebas genéticas de su ADN (o genoma). Algunas variantes tienen importancia clínica y pueden dar a los consumidores información sobre cómo monitorear su salud, o sobre posibles enfermedades o afecciones.
- No todas las pruebas directas al consumidor son pruebas genéticas, aunque la mayoría en el mercado hoy en día lo son. Algunos miden otras cosas, como los tipos de flora bacteriana (referida como un “microbioma”).
- Las pruebas directas al consumidor tienen diferentes niveles de evidencia que respaldan sus afirmaciones. Algunas pruebas directas al consumidor tienen muchos datos científicos y clínicos para respaldar la información que proporcionan, mientras que otras pruebas no tienen tantos datos de apoyo
- Pruebas basadas en ácidos nucleicos: las pruebas analizan variaciones en la secuencia, estructura o expresión del ácido desoxirribonucleico (ADN) y ácido ribonucleico (ARN) para diagnosticar enfermedades o afecciones médicas, infección con un patógeno identificable o determinar el estado de portador genético. https://www.fda.gov/medical-devices/vitro-diagnostics/nucleic-acid-based-tests
- Prueba desarrollada en laboratorio (LDT): Una prueba desarrollada en laboratorio (LDT) es un tipo de prueba diagnóstica in vitro diseñada, fabricada y utilizada dentro de un solo laboratorio. Los LDT pueden ser utilizados para medir o detectar una amplia variedad de analitos (sustancias como proteínas, compuestos químicos como glucosa o colesterol, o ADN), en una muestra tomada de un cuerpo humano” (FDA.gov). https://www.fda.gov/medical-devices/vitro-diagnostics/laboratory-developed-tests
- Medicina de Precisión: La mayoría de los tratamientos médicos están diseñados para el “paciente promedio” como un enfoque único para todos, lo que puede ser exitoso para algunos pacientes pero no para otros. La medicina de precisión, a veces conocida como “medicina personalizada”, es un enfoque innovador para adaptar la prevención y el tratamiento de enfermedades que considera las diferencias en los genes, entornos y estilos de vida de las personas. El objetivo de la medicina de precisión es orientar los tratamientos adecuados a los pacientes adecuados en el momento adecuado. https://www.fda.gov/medical-devices/vitro-diagnostics/precision-medicine
Los avances en la medicina de precisión ya han llevado a poderosos descubrimientos y tratamientos aprobados por la FDA que se adaptan a características específicas de los individuos, como la composición genética de una persona o el perfil genético del tumor de un individuo. Los pacientes con una variedad de cánceres se someten rutinariamente a pruebas moleculares como parte de la atención al paciente, lo que permite a los médicos seleccionar tratamientos que mejoran las posibilidades de supervivencia y reducen la exposición a efectos adversos.
Pruebas de Secuenciación de Próxima Generación (NGS)
La atención de precisión solo será tan buena como las pruebas que guían el diagnóstico y el tratamiento. Las pruebas de Secuenciación de Próxima Generación (NGS) son capaces de identificar rápidamente o 'secuenciar' grandes secciones del genoma de una persona y son avances importantes en las aplicaciones clínicas de la medicina de precisión. Pacientes, médicos e investigadores pueden usar estas pruebas para encontrar variantes genéticas que les ayuden a diagnosticar, tratar y comprender más sobre las enfermedades humanas.
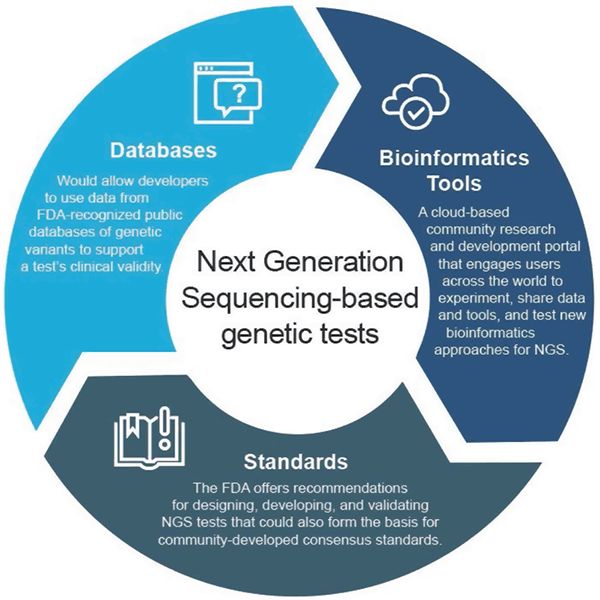
Plataforma Bioinformática de la FDA
La FDA creó https://precision.fda.gov/, un portal de investigación y desarrollo comunitario basado en la nube que involucra a usuarios de todo el mundo para compartir datos y herramientas para probar, probar y validar el enfoque bioinformático existente y nuevo del procesamiento de NGS. Los individuos y organizaciones de la comunidad genómica pueden encontrar más información e inscribirse para participar en http://precision.fda.gov.
Solo para uso en investigación (RUO) y solo para uso en investigación (IUO)
Ambos IVD se consideran precomerciales ya que no se utilizan con fines diagnósticos y no tienen que seguir los estrictos requisitos de etiquetado que se aplican a los IVD de diagnóstico comercial. Las necesidades de etiquetado de estos se encuentran en 21 CFR 809.10. La diferencia entre RUO e IUO es que RUO es solo para investigación, pero la IUO puede ser enviada previamente y puede evaluarse para su uso futuro como DIV.
Reactivos de propósito general (GPR) y reactivos específicos para analitos (ASR)
GPR tiene una aplicación general de laboratorio y es un dispositivo de Clase I. Como tal, los dispositivos Clase I están exentos de PMN. El ASR es un poco más complicado ya que puede usarse como dispositivo Clase I, II o III dependiendo de su aplicación. Los dispositivos ASR pueden variar desde anticuerpos a proteínas de unión a ácido nucleico utilizadas para el diagnóstico en muestras de bancos de sangre (clase II) hasta una prueba para el Ébola (Clase III).
Enmiendas a la mejora de laboratorio clínico (CLIA)
Las pruebas de diagnóstico ayudan a los proveedores de atención médica a detectar o monitorear enfermedades o afecciones específicas. También ayuda a evaluar la salud del paciente para tomar decisiones clínicas para la atención al paciente. Las Enmiendas a la Mejora del Laboratorio Clínico (CLIA) regulan las pruebas de laboratorio y requieren que los laboratorios clínicos sean certificados por su estado, así como por el Centro de Servicios de Medicare y Medicaid (CMS) antes de que puedan aceptar muestras humanas para pruebas de diagnóstico. Los laboratorios pueden obtener múltiples tipos de certificados CLIA, basados en los tipos de pruebas diagnósticas que realizan. www.fda.gov/medical-devices/ivd-regulatoryassistance/clinical-laboratory-improvement-amendments-clia
En 1988, se establecieron las Enmiendas de Mejoramiento de Laboratorio Clínico (CLIA) para definir estándares de calidad para todas las pruebas de laboratorio para garantizar la precisión, confiabilidad y puntualidad de los resultados de las pruebas de los pacientes independientemente de dónde se realizó la prueba. El reglamento definitivo se estableció en 1992. Tres agencias federales son responsables de CLIA: la Administración de Alimentos y Medicamentos (FDA), el Centro de Servicios de Medicaid (CMS) y el Centro para el Control de Enfermedades (CDC). Cada agencia tiene un papel único en asegurar pruebas de laboratorio de calidad. www.cms.gov/regulations-andguidance/legislation/clia/program_descriptions_projects.html
CLIA Y LA FDA, CMS, CDC
Tres agencias federales son responsables de CLIA: la Administración de Alimentos y Medicamentos (FDA), el Centro de Servicios de Medicaid (CMS) y el Centro para el Control de Enfermedades (CDC). Cada agencia tiene un papel único en asegurar pruebas de laboratorio de calidad.
Categorizaciones CLIA
La FDA clasifica las pruebas diagnósticas por su complejidad, desde las menos a las más complejas: pruebas exentas, pruebas de complejidad moderada y pruebas de alta complejidad. Las pruebas diagnósticas se clasifican como renunciadas con base en la premisa de que son simples de usar, y hay pocas posibilidades de que la prueba proporcione información incorrecta o cause daño si se hace incorrectamente. A las pruebas aprobadas por la FDA para uso doméstico o de venta libre se les asigna automáticamente una categorización exenta.
La categorización CLIA se determina después de que la FDA haya aprobado o aprobado una presentación de comercialización. La FDA determina la complejidad de la prueba revisando las instrucciones de prueba del prospecto y usando un “cuadro de mando” de criterios para categorizar una prueba como de complejidad moderada o alta. Cada prueba se califica por nivel de complejidad asignando puntajes de 1, 2 o 3 para cada uno de los siete criterios del cuadro de mando.
Una puntuación de 1 indica el nivel más bajo de complejidad, y el puntaje de 3 indica el nivel más alto. Las 7 puntuaciones se suman y las pruebas con una puntuación de 12 o menos se clasifican como de complejidad moderada, y las que tienen una puntuación superior a 12 se clasifican como de alta complejidad. La FDA notificará al patrocinador, generalmente dentro de las dos semanas posteriores a la autorización de comercialización o aprobación de su categorización CLIA.
Conoce más aquí: https://www.fda.gov/medical-devices/ivd-regulatory-assistance/clia-categorizations
Solicitud de Exención CLIA
Bajo CLIA, la FDA clasifica las pruebas de diagnóstico in vitro (IVD) por su grado de complejidad: renunciado, complejidad moderada y alta complejidad. Las pruebas que no son eximidas por la regulación bajo 42 CFR 493.15 (c), o aprobadas o aprobadas para uso doméstico o para uso de venta libre, se clasifican automáticamente como renunciadas después de la autorización o aprobación. De lo contrario, después de la autorización o aprobación, las pruebas pueden clasificarse como de complejidad moderada o alta de acuerdo con los criterios de categorización CLIA enumerados en 42 CFR 493.17.
El estatuto establece que: Los exámenes y procedimientos que pueda realizar un laboratorio con Certificado de Exoneración son exámenes y procedimientos de laboratorio que hayan sido aprobados por la Administración de Alimentos y Medicamentos para uso doméstico o que sean simples exámenes y procedimientos de laboratorio que cuenten con un riesgo insignificante de un resultado erróneo, incluyendo aquellos que — (A) emplean metodologías que son tan simples y precisas que hacen que la probabilidad de resultados erróneos por parte del usuario sea insignificante, o (B) el Secretario ha determinado que no plantean riesgo irrazonable de daño al paciente si se realiza incorrectamente.
¡Explora!
Hay tres formas de averiguar qué categorización ha recibido una prueba de laboratorio. Echa un vistazo a estas tres bases de datos.
- Base de datos CLIA
- Analitos Renunciados
- Base de datos sobre el mostrador