
10.3: Actividades de cumplimiento
- Page ID
- 53020

Ejecución Civil
Las regulaciones deben ser ejecutables para ser efectivas, y la FDA tiene muchas herramientas para fomentar el cumplimiento. La clave de la FDA es la salud pública y la seguridad del producto. Los procedimientos de ejecución suelen tener lugar después de que se haya considerado OAI una inspección. Las inspecciones, a través de los centros correspondientes, pueden ser ya sea cada dos años o como lo indique un tema del que se haya informado a la FDA. Existen dos amplias categorías de actividades de ejecución; civil y penal.
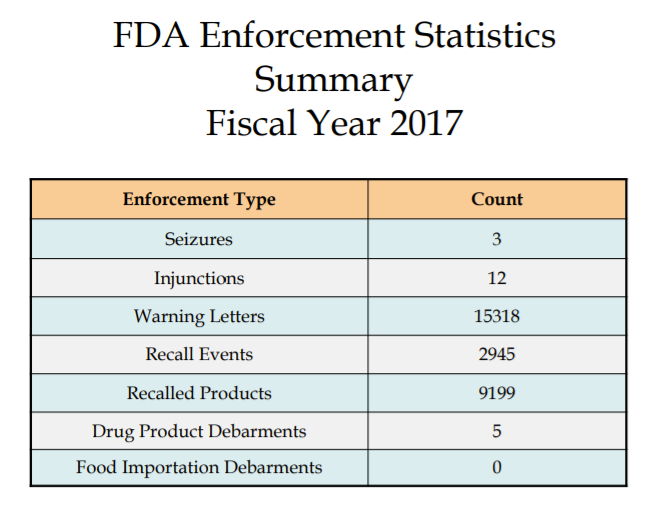
Los lineamientos para las acciones de cumplimiento que toma la FDA se pueden encontrar en la página web de Inspecciones, Cumplimiento, Aplicación e Investigación Criminal (ICECI). La FDA publica actividades de cumplimiento en informes anuales en su sitio web. Mire a su alrededor y vea qué tipo de actividades de aplicación son las más destacadas. Las estadísticas de 2017 se pueden encontrar aquí: https://www.fda.gov/downloads/ICECI/EnforcementActions/UCM592790.pdf
Inspecciones
La FDA tiene la autoridad legal para inspeccionar a las compañías farmacéuticas, y puede hacerlo sin previo aviso. Los lineamientos para las inspecciones se publican en la página web de Inspecciones, Cumplimiento, Ejecución e Investigación Criminal (ICECI). Diferentes pautas de inspección rigen diferentes tipos de productos. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references
- Manual de Operaciones de Inspecciones: www.fda.gov/inspecciones-cumplimiento-y-investigaciones-criminales/referencias-inspecciones/investigaciones-operaciones-manual
- Guías de inspección: www.fda.gov/inspecciones-cumplimiento-y-investigaciones-criminales/referencias-inspecciones/guías de inspección
- Guías de Inspección Extranjera: www.fda.gov/inspecciones-cumplimiento-y-investigaciones-criminales/referencias-inspecciones/inspecciones-inspecciones/inspecciones extranjeras
La FDA realiza inspecciones de rutina (cada dos años) para cumplir con CGMP, GLP, GCP y Regulaciones del Sistema de Calidad. También realizan inspecciones específicas basadas en otros eventos de aplicación, como seguimientos de eventos de retiro o cartas de advertencia. Cuando la FDA ingresa a una instalación para su inspección, proporcionan el documento de solicitud de inspección (FDA 482) así como proporcionan identificación. Si se les niega la entrada, la FDA puede obtener una orden de cateo de un tribunal federal.
El alcance de la inspección se basa principalmente en el motivo de la visita y el historial de la instalación. Los únicos límites que tiene la FDA en las inspecciones se refieren a la información financiera, los datos de ventas (no relacionados con el envío), los datos de personal (no relacionados con la capacitación) y los datos de investigación (no relacionados con la aprobación de productos).
Durante las inspecciones, la FDA analiza siete sistemas para determinar si están 'en control':
- Responsabilidad Gerencial
- Control de diseño
- Acción correctiva y preventiva (CAPA)
- Controles de producción y procesos
- Registros y controles de cambio de documentos
- Controles de materiales
- Controles de instalaciones y equipos
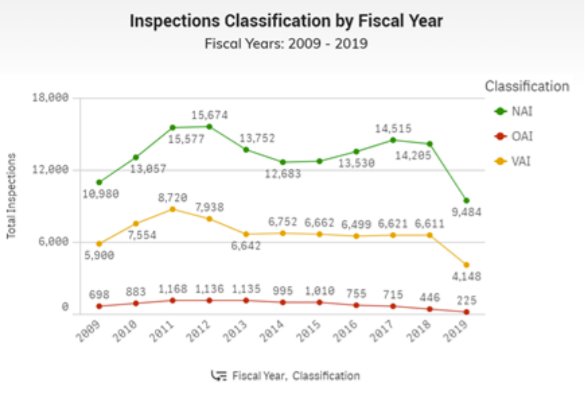
Clasificación de inspección
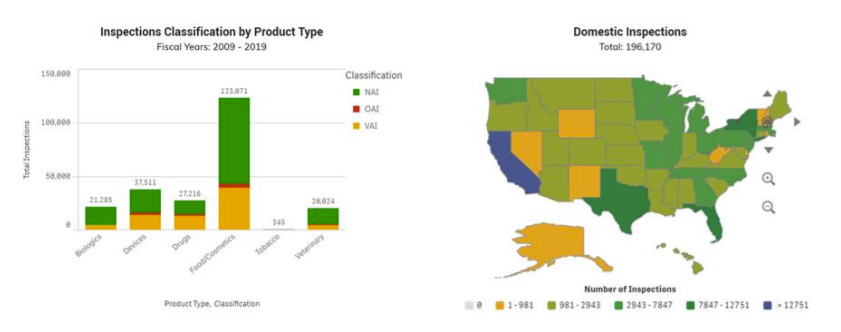
Después de una inspección, la FDA determina si las áreas evaluadas cumplen con las leyes y regulaciones aplicables. La FDA y la Base de Datos de Clasificación de Inspección clasifican la inspección por cada área del proyecto con una de tres clasificaciones. La Base de Datos de Clasificación de Inspección solo muestra las inspecciones realizadas por la FDA. www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-classification-database
La FDA realiza inspecciones cuidadosas de las instalaciones que realizan estudios de laboratorio no clínicos para determinar el cumplimiento de la Parte 58 (Buenas Prácticas de Laboratorio para Estudios de Laboratorio No Clínicos) del Título 21 del CFR. Los estudios de laboratorio no clínicos son experimentos en los que los artículos de prueba se estudian prospectivamente en sistemas de prueba, como animales, plantas, microorganismos en condiciones de laboratorio para determinar su seguridad. Conoce más aquí: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/nonclinical-laboratories-inspected-under-good-laboratory-practices
Las tres clasificaciones de inspección que se muestran son:
- Ninguna Acción Indicada (NAI) lo que significa que no se encontraron condiciones o prácticas objetables durante la inspección (o las condiciones objetables encontradas no justifican una acción reglamentaria adicional),
- Acción Voluntaria Indicada (VAI) lo que significa que se encontraron condiciones o prácticas objetables, pero el organismo no está preparado para tomar o recomendar ninguna acción administrativa o reglamentaria, o
- Acción Oficial Indicada (OAI), lo que significa que se recomendarán acciones reglamentarias o administrativas.
Citas de inspección: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-citation
Inspecciones y Formulario 483
La adulteración y la falta de marca son las dos violaciones más comunes encontradas durante una inspección. Las instalaciones farmacéuticas y las empresas biotecnológicas son inspeccionadas periódicamente por la FDA. Cualquier violación de CGMP que los inspectores encuentren se anotan en formularios, denominados “483's”, y si no hay respuesta a los temas de cumplimiento normativo, se envía una Carta de Advertencia oficial a la empresa.
A 483 es resultado de una inspección y aviso de incumplimiento regulatorio. Se trata de una lista de ítems específicos vistos al término de la inspección y es emitida por el investigador. Después de una inspección, el inspector se reúne con el encargado de las instalaciones para abordar tantos de los problemas antes de que se escriba el 483. El inspector ofrece la oportunidad de remediar una situación inmediatamente después de la inspección y documentar el cumplimiento y los recursos, o soluciones y acuerdos futuros. Después de que la carta sea escrita y recibida por la empresa, la empresa cuenta entonces con 15 días hábiles para responder voluntariamente al 483 por escrito con un plan de acción. Si el 483 emitido no se soluciona rápidamente, la FDA puede responder con una carta de advertencia (ver más abajo).
Cabe señalar que la emisión del modelo 483 no significa automáticamente que una firma no esté en cumplimiento de GMP. Adicionalmente, la longitud del formulario no es un indicador confiable de la gravedad de las violaciones observadas. El formulario debe ser visto críticamente, y las empresas que sientan que han recibido un 483 que contiene observaciones cuestionables de desviaciones de GMP deben discutir el tema con el inspector, director de distrito, director regional, o incluso el centro de emisión si es necesario.
No se requiere que las empresas respondan a un 483. Sin embargo, se recomienda su respuesta. Si una empresa recibe demasiados 483 y no proporciona a la FDA respuestas adecuadas a estos formularios, se les somete a una carta de advertencia o a más medidas punitivas como se describe a continuación.
Establecimiento de Informes de Inspección
Después de que la FDA cierra una inspección, publican un Informe de Establecimiento de Inspección (EIR). Se trata de un informe formal escrito que resume los hallazgos con evidencia de apoyo. En este reporte, la FDA clasifica la inspección como No Acción Indicada (NAI), Indicación de Acción Voluntaria (VAI) o Acción Oficial Indicada (OAI). Si se anota una OAI, las actividades de aplicación pueden ser listadas o ser programadas para venir. Informes y datos de inspección: https://datadashboard.fda.gov/ora/cd/inspections.htm
Inspecciones y cartas de advertencia
Cuando la FDA descubre que un fabricante ha violado significativamente las regulaciones de la FDA, la FDA notifica al fabricante, la mayoría de las veces, en forma de una carta de advertencia. La Carta de Advertencia identifica la violación y deja claro que la empresa debe corregir el problema y proporciona instrucciones y un plazo para que la compañía informe a la FDA de sus planes de corrección. Luego, la FDA verifica para asegurarse de que las correcciones de la compañía sean adecuadas.
Los asuntos descritos en las cartas de advertencia de la FDA pueden haber estado sujetos a una interacción posterior entre la FDA y el destinatario de la carta que puede haber cambiado el estado regulatorio de los temas discutidos en la carta. Una carta de advertencia emitida por la FDA es una comunicación voluntaria de asesoría informal y ofrece a una empresa la oportunidad de remediar la situación regulatoria. El objetivo general de una carta de advertencia es fomentar el cumplimiento voluntario. Si el tema no pone en peligro inmediato al público, se podrá escribir una carta de advertencia antes de otras medidas punitivas. Un aspecto importante de una carta de advertencia es establecer un registro de 'previo aviso' durante los procedimientos legales. Sin embargo, cuando se emite, una carta de advertencia se publica inmediatamente en el sitio web de la FDA y se comparte entre las agencias reguladoras.
- Ver cartas de advertencia generales de la FDA: www.fda.gov/inspecciones-cumplimiento-cumplimiento-investigaciones-criminales/cumplimiento-acciones-y-actividades/cartas de advertencia
- Ver cartas de advertencia de la FDA sobre tabaco: www.fda.gov/inspecciones-cumplimiento-cumplimiento-investigaciones-criminales/cartas-advertencias/tabaco-minoristas de advertencia-cartas
- Ver cartas de advertencia de marketing y publicidad de medicamentos: www.fda.gov/medicamentos/aplicaciones-actividades-fda/advertencia-cartas-y-notar-violacion-letras-empresas-farmacéuticas
Terminaciones de cartas de advertencia
La FDA puede emitir una carta de cierre de carta de advertencia una vez que hayan completado una evaluación y hayan determinado que una empresa emprendió acciones correctivas en respuesta a una carta de advertencia. Las acciones correctivas deben haber sido realizadas y verificadas por la FDA, generalmente a través de una inspección de seguimiento. Si la Carta de Advertencia contiene violaciones que por su naturaleza no son corregibles, entonces no se emitirá ninguna carta de cierre. Las futuras inspecciones de la FDA y las actividades regulatorias pueden evaluar aún más la idoneidad y sostenibilidad de estas correcciones. En caso de que se observen violaciones durante una inspección posterior o por otros medios, se podrán tomar medidas coercitivas sin previo aviso.
Inspecciones y cartas sin título
Las cartas sin título son la forma menos dura de comunicación con la FDA. Al igual que las cartas de advertencia, las cartas sin título pueden notificar a una empresa de una violación que puede no alcanzar el umbral de un problema regulatorio. A diferencia de una carta de advertencia, una carta sin título no incluye una declaración que advierta a la persona o empresa de que el hecho de no corregir rápidamente la violación puede resultar en acciones ejecutivas. F Sin embargo, se anima a las empresas a abordar cualquier problema ya que esto sí constituye una 'notificación previa' y es por eso que debe responder. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/issuance-untitled-letters. Para más información sobre advertencia y cartas sin título: https://www.fda.gov/media/71878/download
Inspecciones y comunicados de prensa
La FDA también puede emitir comunicados de prensa desfavorables o avisos del registro federal. En caso de un defecto atroz, la FDA puede procesar penalmente, incautar materiales y ejecutar medidas cautelares contra una empresa y particulares. (fda.gov). Comunicados de prensa actuales: www.fda.gov/inspecciones-cumplimiento-aplicacion-investigaciones-criminales/investigaciones-criminales/comunicados de prensa
Convulsiones
Si una empresa se niega a retirar el mercado, la FDA puede presentar un caso de incautación o orden judicial en su contra para abordar las violaciones incluso si los productos no son defectuosos. Entonces, pueden solicitar al tribunal una orden que permita a los funcionarios federales tomar posesión de drogas “adulteradas” y destruirlas. Este proceso permite a la FDA evitar inmediatamente que una compañía distribuya medicamentos defectuosos y potencialmente dañinos a los consumidores. Tanto los casos de incautación como los de orden judicial suelen dar lugar a órdenes judiciales que requieren que las empresas tomen muchas medidas para corregir las violaciones. Estos pasos pueden incluir la contratación de expertos externos para ayudar a resolver el problema, redactar nuevos procedimientos y capacitar a los empleados. En algunos casos, las violaciones pueden convertirse en casos penales, lo que permite a la FDA buscar multas y tiempo en la cárcel.
Mandaturas
Si una empresa viola la Ley FD&C, la FDA puede presentar una orden judicial contra la empresa. Esta orden judicial es un proceso judicial civil y generalmente se usa cuando se identifica un peligro significativo para la salud con un producto. Esta FDA puede solicitar una orden de restricción temporal, una orden judicial temporal o una orden judicial permanente. La clave aquí es que la FDA pueda actuar rápidamente para que un producto deje de llegar a los clientes a falta de un retiro del mercado (que la FDA no tiene autoridad en algunos casos). La FDA también utiliza este método de aplicación si la compañía ha ignorado repetidas cartas de advertencia. Si la empresa aborda los temas, se puede levantar la orden de restricción temporal.
Sanciones civiles monetarias
La Ley FD&C, así como la PHSA, tienen disposiciones civiles de penalización monetaria. La orientación sobre CMP se proporciona en 21 CFR 17.2. ¡Busque el CFR en el sitio web de la FDA y observe que los CMP son bastante duros! Algunas penas superan el millón de dólares por delitos agregados.
Descalificación de Investigadores Clínicos
La FDA regula los estudios científicos diseñados para desarrollar evidencia que respalde la seguridad y efectividad de los medicamentos en investigación (humanos y animales), productos biológicos y dispositivos médicos. Los médicos y otros expertos calificados (“investigadores clínicos”) que realicen estos estudios están obligados a cumplir con los estatutos y regulaciones aplicables destinados a garantizar la integridad de los datos clínicos en los que se basan las aprobaciones de productos y, en el caso de investigaciones que involucren a sujetos humanos, ayudar a proteger los derechos, la seguridad y el bienestar de esos sujetos. En ciertas situaciones, en las que la FDA alega que un investigador clínico ha violado las regulaciones aplicables, la FDA puede descalificar a un investigador clínico y evitar que proporcione datos clínicos para cualquier envío de producto. Normalmente, la información de los investigadores clínicos se obtiene a través de la inspección BIMO. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/clinical-investigators-disqualification-proceedings
Sanción
La FDA tiene la autoridad de inhabilitación de individuos o empresas de la industria farmacéutica. La inhabilitación significa que ya no pueden fabricar un medicamento aprobado. Las empresas deshabilitadas ya no pueden fabricar, ni presentar más solicitudes de medicamentos. En 2018, la FDA descartó una entidad corporativa por primera vez. Lee más aquí: https://www.federalregister.gov/documents/2018/03/01/2018-04195/meunerie-sawyerville-inc-denial-of-hearing-final-debarment-order.
Las personas deshabilitadas también pueden estar sujetas a sanciones civiles monetarias, también. Consulte una lista de inhabilitaciones en el sitio web de la FDA. ¡Notarás que es una lista sorprendentemente larga! https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/fda-debarment-list-drug-product-applications
Ejecución Penal
Las investigaciones penales son realizadas por la Oficina de Investigaciones Penales (OCI). Es importante recordar cuando estamos discutiendo las regulaciones de la FDA sobre Alimentos y Medicamentos, nos estamos refiriendo a las leyes. El quebrantamiento de leyes tiene muchas sanciones, que pueden ir desde multas hasta tiempo en la cárcel. Echa un vistazo a los fugitivos más buscados de la FDA aquí: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/criminal-investigations/office-criminal-investigations-most-wanted-fugitives
Decreto de Consentimiento
Las empresas que incumplan reiteradamente los requisitos de CGMP podrán verse obligadas a realizar cambios mediante la emisión de un decreto de consentimiento. El decreto de consentimiento es firmado por el máximo funcionario de la compañía, el Fiscal de los Estados Unidos y el Tribunal de Distrito de Estados Unidos. Posteriormente se presenta el decreto ante el tribunal y posteriormente se somete a la FDA. Aplicados por los tribunales federales, los decretos de consentimiento suelen implicar multas, reembolsos al gobierno por costos de inspección y sanciones por incumplimiento. Los decretos de consentimiento pueden ser permanentes. No obstante, si una empresa ha cumplido, puede solicitar al tribunal que retire el decreto.
¡Pon a prueba tus conocimientos!
Lea aquí una historia destacada en el sitio web de la OCI: “4 de abril de 2016: Ex residente de Carlsbad encarcelado por venta de dispositivos médicos “Energy Wave” no aprobados”.
- ¿Qué es lo que se acusa de hacer a David Pérez?
- ¿Por qué es esto en contra de la ley? ¿Se puede hacer referencia a la ley?
- ¿Cuánto tiempo de sentencia recibió? Si no recibió un acuerdo de declaración de culpabilidad y fue declarado culpable en un tribunal de justicia, ¿cuánto tiempo habría sido su sentencia máxima?
- ¿Está de acuerdo con el cargo y su sentencia? Explique.