
2.2: Microscopio óptico o de luz
- Page ID
- 51400

El término microscopia óptica se refiere al empleo de cualquier clase de microscopio que utilice luz visible para observar las muestras. Según las propiedades luminosas del instrumento existen distintos tipos de microscopios ópticos: de campo claro, contraste de fase, campo oscuro y fluorescencia.
Microscopio de campo claro
Un microscopio de campo claro tiene una serie de lentes y utiliza luz visible como fuente de iluminación (Figura 2.1). Un conjunto de lentes forma una imagen focal definida cuyo tamaño es muchas veces mayor que el de la muestra en sí. Este aumento se logra cuando los rayos luminosos procedentes de la fuente de luz pasan a través de un condensador, que tiene lentes que dirigen los rayos de luz a través de la muestra. Desde aquí los rayos pasan al interior del objetivo, la lente más próxima a la muestra. La imagen de la muestra vuelve a ser ampliada por el ocular.
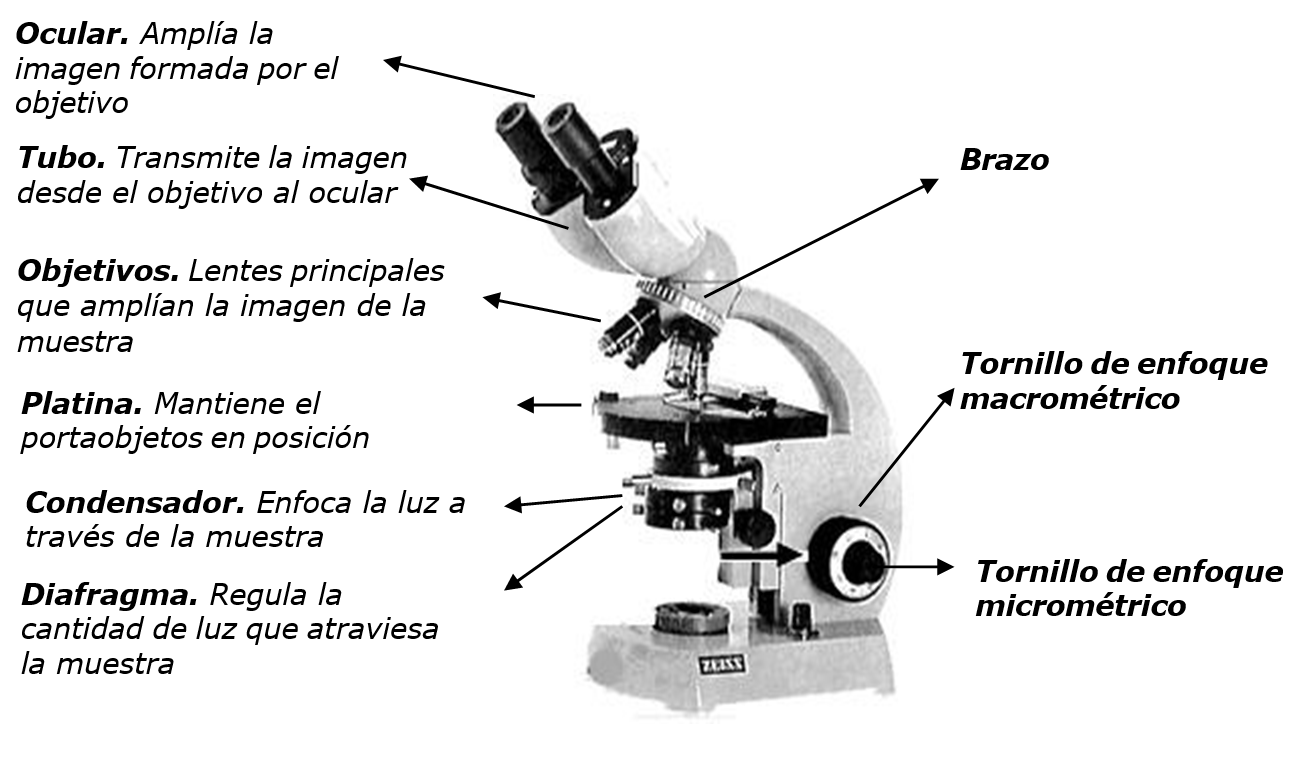
La capacidad amplificadora de un microscopio compuesto es el producto del aumento individual de los oculares y los lentes objetivos. La mayoría de los microscopios utilizados en microbiología poseen varias lentes objetivos, que proporcionan 10x (bajo aumento), 40x (gran aumento) y 100x (de inmersión en aceite). La mayoría de los oculares amplían la imagen 10 veces. Al multiplicar el aumento de un objetivo específico por el del ocular (10x) se observa que el aumento total puede ser de 100A con bajo aumento, de 400A con gran aumento y 1000A con lente de inmersión.
La resolución se define como el espacio de máxima aproximación entre dos puntos en el que aún se pueden observar claramente como dos entidades independientes. El poder de resolución de un microscopio está sujeto a la longitud de onda de la luz y a la propiedad de las lentes conocidas como la apertura numérica (AN). El límite del poder de resolución de un microscopio es aproximadamente igual a 0,61/AN que para un microscopio óptico es de alrededor de 200 nm.
A menor longitud de onda de la luz y AN de las lentes, el poder de resolución del microscopio será mejor. Por lo tanto, queda claro que el poder de resolución de los microscopios ópticos se encuentra restringido por la AN que se obtenga de los sistemas de lentes y las longitudes de onda del espectro de luz visible.
La apertura numérica indica la cantidad de luz que entra en un objetivo desde un punto del campo del microscopio. Tal valor es muy importante, ya que, de él depende el poder de resolución, la propiedad más importante de un objetivo.
La AN de una lente depende del índice de refracción (N) del medio que llena el espacio entre el objeto y la parte frontal del objetivo, y del ángulo (μ) de los rayos de luz más oblicuos que puedan entrar al objetivo. La fórmula para calcular la AN es: AN = N x sen μ/2.
El aire tiene un índice de refracción de 1, que limita la resolución que se puede obtener, pero se puede incrementar la AN poniendo aceite de inmersión entre el espécimen y el objetivo, aumentando así el poder de resolución del microscopio. El aceite de inmersión tiene un índice de refracción de 1,5 lo cual aumenta considerablemente la AN y esto mejora el poder de resolución del microscopio.
Para obtener una imagen clara con detalles precisos en el microscopio óptico compuesto las muestras deben teñirse para que contrasten nítidamente con el medio que las rodea. Para lograr ese contraste es necesario cambiar el índice de refracción de la muestra al del medio, esto se logra tiñendo las muestras. En las condiciones habituales de trabajo el campo de visión de un microscopio óptico está iluminado de forma brillante. Cuando se enfoca la luz, el condensador produce una iluminación brillante (campo claro).
Microscopio de campo oscuro
Se utiliza para examinar microorganismos vivos que no son visibles con el microscopio óptico común, que no pueden teñirse con los métodos estándares o que resultan tan distorsionados por la tinción que sus características no pueden identificarse. El sistema de iluminación está modificado de tal modo que la luz incide sobre la muestra sólo desde los lados. La única luz que es capaz de entrar en el objetivo es la que es dispersada por la muestra y, por lo tanto, los microorganismos se observan brillantes sobre un fondo oscuro. La resolución de dichos microscopios es bastante alta y pueden observarse en ellos objetos difícilmente percibidos por microscopía de campo claro o de contraste de fases. Se utiliza para examinar microorganismos no teñidos suspendidos en un líquido, por ejemplo, espiroquetas muy delgadas como Treponema pallidum, el agente causal de la sífilis.
Microscopio de contraste de fases
Permite la observación detallada de estructura interna, cápsulas, endosporas, gránulos citoplasmáticos y movilidad en los microorganismos vivos. Además no es necesario fijar (adherir los microorganismos al portaobjetos) ni teñir las muestras, procedimientos que podrían distorsionar o destruir a los microorganismos.
Este tipo de microscopia se basa en el hecho de que las células poseen un índice de refracción distinto al del medio y por lo tanto desvían los rayos de luz que las atraviesan. La luz que pasa a través de la muestra con índice de refracción diferente al del medio que la rodea sufre un cierto retardo. Este efecto es amplificado por un anillo especial que posee el objetivo, lo que da lugar a la formación de una imagen oscura sobre un fondo brillante.
Microscopio de contraste por interferencia diferencial (mcid)
Es similar al microscopio de contraste de fase en que utiliza diferencias en los índices de refracción. Sin embargo, un MCID emplea dos haces de luz en lugar de uno. Además los prismas dividen cada haz, lo que agrega colores contrastantes a la muestra. Por consiguiente, la resolución de un MCID es mayor que la del microscopio de contraste de fase estándar. Por otra parte, la imagen tiene colores brillantes y parece casi tridimensional.
Microscopio de fluorescencia
Se utiliza para visualizar muestras capaces de emitir fluorescencia, es decir, capaces de emitir una determinada longitud de onda cuando previamente ha incidido sobre ellas una luz de menos longitud de onda (ultravioleta). La fluorescencia puede ser debida a que determinadas células poseen sustancias fluorescentes naturales como la clorofila o por haber sido previamente tratadas con un colorante fluorescente (fluorocromo).
La auramina O brilla con un color amarillo cuando se expone a la luz UV, es fuertemente absorbido por Mycobacterium tuberculosis; Bacillus anthracis aparece de color verde manzana cuando se lo tiñe con isotiocianato de fluoresceína (FITC). Estas técnicas son muy usadas en Ecología Microbiana y en diferentes técnicas de base Inmunológica dentro de su disciplina y en diferentes áreas de la Microbiología.
Una de las limitaciones de los tipos de microscopía óptica que se han descrito anteriormente es que las imágenes que se obtienen son esencialmente bidimensionales. Sin embargo, esta limitación se puede superar utilizando otros tipos de microscopios.
Microscopio confocal
La microscopía confocal (microscopía laser confocal de barrido) presenta algunas ventajas sobre la microscopía óptica convencional, como son su mayor contraste y elevada resolución, especialmente en aquellas células marcadas con fluorescencia. En estas células se incrementa notablemente la resolución axial (profundidad), permitiendo el seccionamiento óptico celular. De esta manera, es posible obtener representaciones tridimensionales celulares. Además, debido a que su fuente de iluminación es un láser, es posible localizar y observar conjuntamente distintos marcadores fluorescentes en la misma célula. Si la comparamos con la microscopía electrónica de transmisión, la microscopía confocal también puede proporcionar información relevante sobre la organización intracelular, aunque los aumentos son claramente inferiores. Las desventajas principales de la microscopía confocal radican en el elevado costo de la instrumentación requerida y la necesidad de personal especializado para su manejo.
Las aplicaciones de la microscopía confocal en el campo de la observación bacteriana son muy diversas. Una de las principales es el estudio de las comunidades microbianas presentes en biopelículas microbianas (placa dental, tapetes microbianos, etc.) y aquellas que conforman la estructura biológica de los flóculos de lodos activos. La microscopía confocal con análisis espectrofotométrico puntual permite la identificación de grupos bacterianos, sin la necesidad de utilización de sondas fluorescentes específicas, cuando las células presentan autofluorescencia. Mediante un análisis de espectro de absorción (excitación) y emisión a distintas longitudes de onda, se puede realizar una caracterización espectrofotométrica “in situ” de la presencia de un determinado pigmento fotosensible en una célula. Mediante una deducción, razonamiento similar a la de los estudios quimiotaxonómicos que se realizan con cultivos bacterianos, podemos asociar la presencia de un grupo bacteriano con la existencia de una determinada molécula en la célula. Por ejemplo, la determinación de una ficobiliproteína (ficocianina, aloficocianina, ficoeritrina) en el interior de una bacteria, indicaría que se trata de una cianobacteria.