6.2: Átomos a Moléculas
- Page ID
- 84204

Ahora buscamos determinar los estados electrónicos de moléculas enteras —orbitales moleculares. Aunque comenzaremos con moléculas relativamente pequeñas, las técnicas de cálculo que introduzcamos se pueden extender a materiales más grandes que generalmente no pensamos como moléculas: como los cristales de Si, por ejemplo.
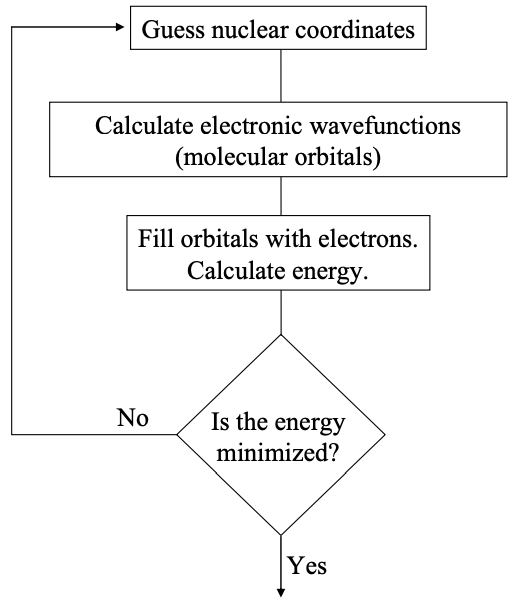
En la discusión previa sobre los orbitales atómicos, asumimos implícitamente que el núcleo es estacionario. Este es un ejemplo de la aproximación Born-Oppenheimer, que señala que la masa del electrón,\(m_{e}\), es mucho menor que la masa del núcleo,\(m_{N}\). En consecuencia, los electrones responden casi instantáneamente a los cambios en las coordenadas nucleares.
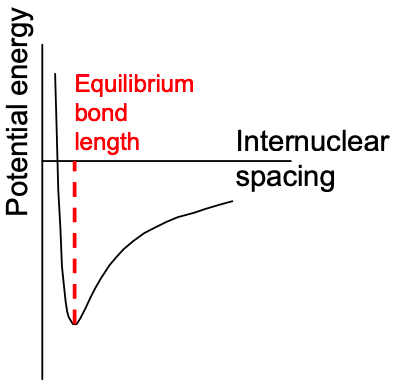
En los cálculos de la estructura electrónica de las moléculas, tenemos que considerar múltiples electrones y múltiples núcleos. Podemos simplificar considerablemente el cálculo asumiendo que las posiciones nucleares son fijas. Entonces se resuelve la ecuación de Schrödinger para los electrones en un potencial estático; ver Apéndice 3. Se eligen diferentes arreglos de los núcleos y se optimiza la solución.