
4.2: La espectrometría infrarroja
- Page ID
- 2337

Los enlaces covalentes en las moléculas orgánicas no son palos rígidos - más bien, se comportan más como resortes. A temperatura ambiente, moléculas orgánicas están siempre en movimiento, ya que sus lazos se extienden, doblan, y tuercen. Estas vibraciones complejas pueden dividirse matemáticamente en modos vibracionales individuales, algunos de los cuales se ilustra a continuación.
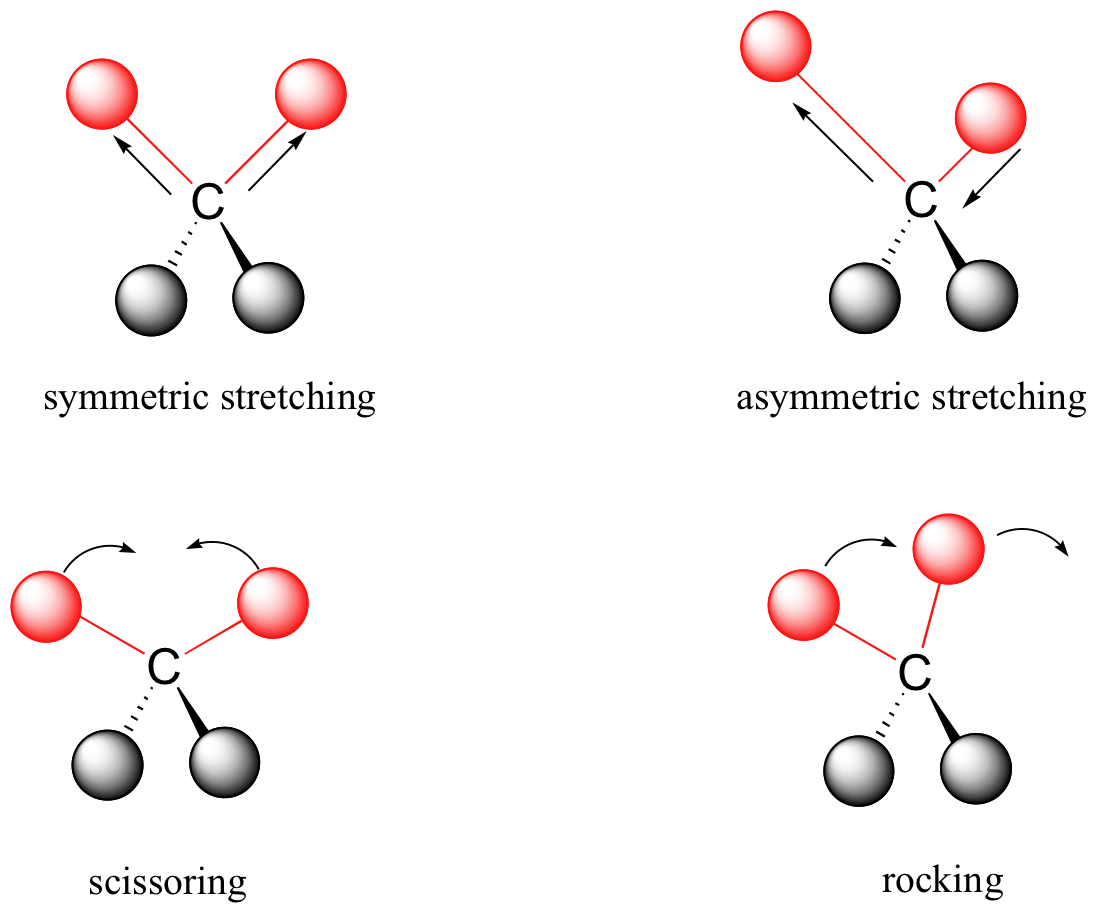
La energía de vibración molecular está cuantizada en lugar de continua, lo que significa que una molécula sólo puede estirar y doblar a ciertas frecuencias 'permitidas'. Si una molécula es expuesta a la radiación electromagnética que coincide con la frecuencia de uno de sus modos de vibración, en la mayoría de los casos la molécula absorberá la energía de la radiación y saltará a un estado de energía de vibración más alta - lo que esto significa es que la amplitud de la vibración aumentará, pero la frecuencia vibracional se mantendrá igual. La diferencia en energía entre los dos estados de vibración equivale a la energía asociada con la longitud de onda de la radiación que fue absorbida. Resulta que es la región infrarroja del espectro electromagnético que contiene frecuencias correspondientes a las frecuencias vibratorias de bonos orgánicos.
Tomemos 2-hexanona como ejemplo. Imagínese el enlace carbonilo del grupo cetona como un resorte. Este resorte está constantemente rebotando de aquí para aya, estirando y comprimiendo, empujando los átomos de carbono y de oxígeno más separados y después jalando los juntos. Este es el modo de estiramiento del enlace carbonilo. En el espacio de un segundo, el resorte 'rebota' de aquí para aya 5.15 x 1013 veces - en otras palabras, la frecuencia del estado fundamental del estiramiento de carbonilo por un grupo de cetona es aproximadamente 5.15 x 1013 Hz.
Si nuestra muestra de cetona se irradia con luz infrarroja, el enlace carbonilo absorberá específicamente la luz con esta misma frecuencia, que por las ecuaciones 4.1 y 4.2 corresponde a una longitud de onda de 5.83 x 10-6 m y una energía de 4.91 kcal/mol. Cuando el enlace carbonilo absorbe esta energía, salta a un estado vibracional excitado.
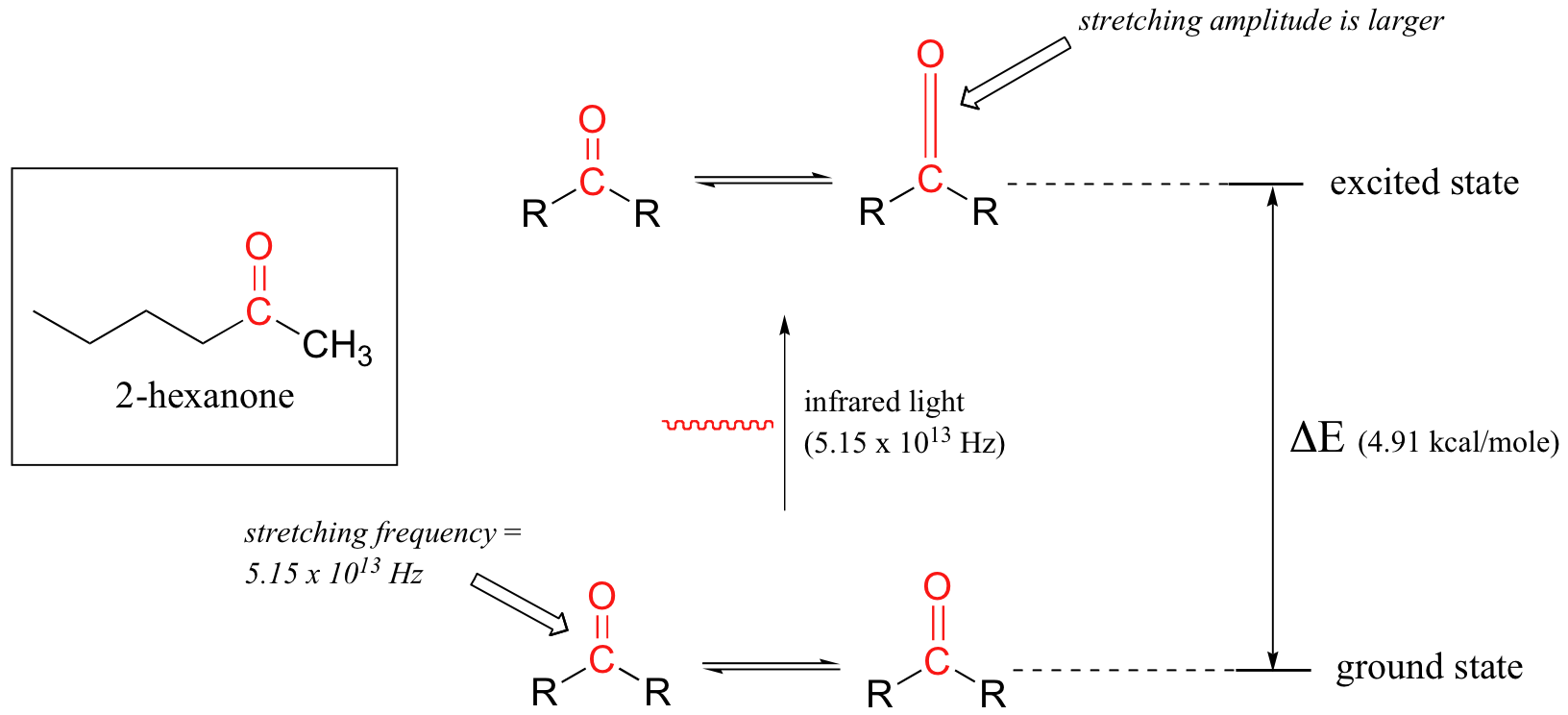
El valor de ΔE - la diferencia de energía entre los estados vibracionales de la energía baja (fundamental) y alta energía (excitado) - equivale a 4.91 kcal/mol, igual a la energía asociada con la frecuencia de luz absorbida. La molécula no permanece en su estado vibracional excitado por mucho tiempo, pero libera rápidamente la energía al ambiente que lo rodea en forma de calor, y devuelve al estado fundamental.
Con un instrumento llamado espectrofotómetro infrarrojo, podemos 'ver' esta transición vibracional. En el espectrofotómetro, luz infrarroja con frecuencias que van desde aproximadamente 1013 a 1014 Hz se pasa a través de nuestra muestra de ciclohexano. La mayoría de las frecuencias pasan a través de la muestra y son registradas por un detector en el otro lado.
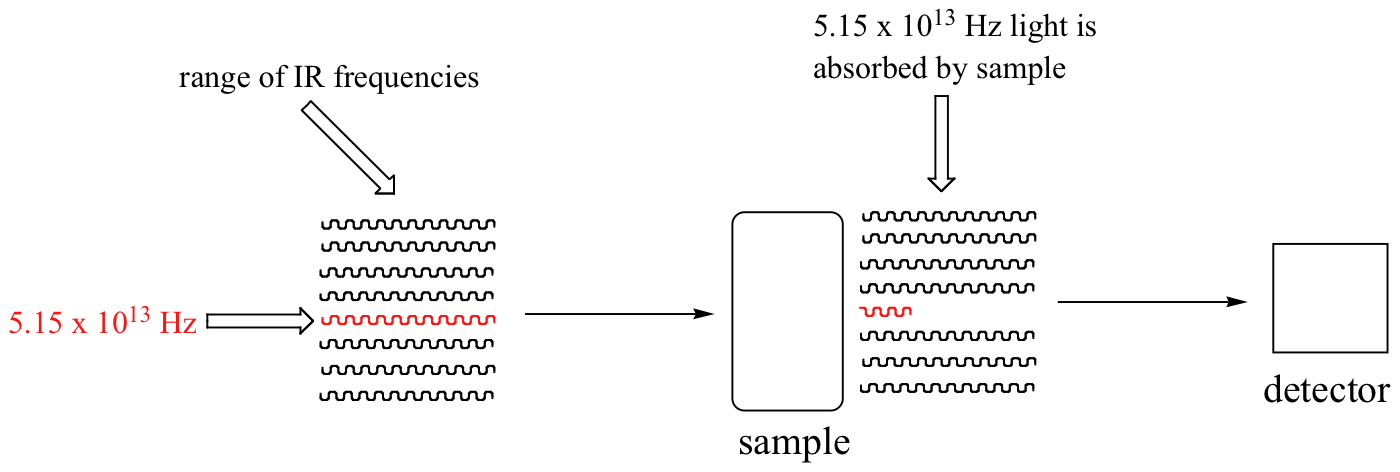
Nuestra 5.15 x 1013 Hz frecuencia de estiramiento de carbonilo, sin emabargo, es absorbida por la muestra 2-hexanona, y por lo que el detector registra que la intensidad de esta frecuencia, después de haber pasado a través de la muestra, es algo menos que 100% de su intensidad inicial.
Las vibraciones de una molécula de 2-hexanona no son, por supuesto, limitadas al simple estiramiento del enlace carbonilo. Los diversos enlaces carbono-carbono también se estiran y doblan, al igual que los enlaces carbono-hidrógeno, y todos estos modos de vibración también absorben diferentes frecuencias de luz infrarroja.
El poder de la espectroscopia infrarroja surge de la observación de que diferentes grupos funcionales tienen diferentes frecuencias de absorción características. El enlace carbonilo en una cetona, como vimos en nuestro ejemplo de 2-hexanona, típicamente absorbe en el rango de 5.11 - 5.18 x 1013 Hz, dependiendo de la molécula. El carbono-carbono triple enlace de un alquino, por otro lado, absorbe en el rango de 6.30 a 6.80 x 1013 Hz. Por tanto, la técnica es muy útil como un medio para identificar qué grupos funcionales están presentes en una molécula de interés. Si pasamos luz infrarroja a través de una muestra desconocida y vemos que absorbe en el rango de frecuencia de carbonilo, pero no en el rango alquino, podemos inferir que la molécula contiene un grupo carbonilo, pero no un alquino.
Algunos bonos absorben la luz infrarroja con más fuerza que otros, y algunos bonos no absorben en lo absoluto. Para que un modo vibracional absorba la luz infrarroja, debe resultar en un cambio periódico en el momento dipolar de la molécula. Tales vibraciones se dicen estar infrarrojo activos. En general, cuanto mayor es la polaridad del enlace, más fuerte será su absorción IR. El enlace carbonílico es muy polar, y absorbe con mucha fuerza. El carbono-carbono triple enlace en la mayoría de los alquinos, por el contrario, es mucho menos polar, y por lo tanto una vibración de estiramiento no da como resultado un gran cambio en el momento dipolar global de la molécula. Grupos alquino absorben más bien débilmente en comparación con carbonilos.
Algunos tipos de vibraciones son infrarrojo inactivos. Las vibraciones de estiramiento de los enlaces dobles y triples completamente simétricos, por ejemplo, no dan lugar a un cambio en el momento dipolar, y por lo tanto no dan lugar a ninguna absorción de luz (pero otros bonos y modos de vibración en estas moléculas sí absorben luz IR).
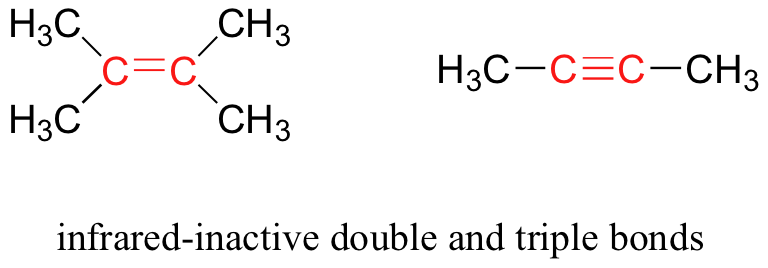
Ahora, miremos algunos datos reales de experimentos de espectroscopía IR. A continuación se muestra el espectro IR de 2-hexanona.
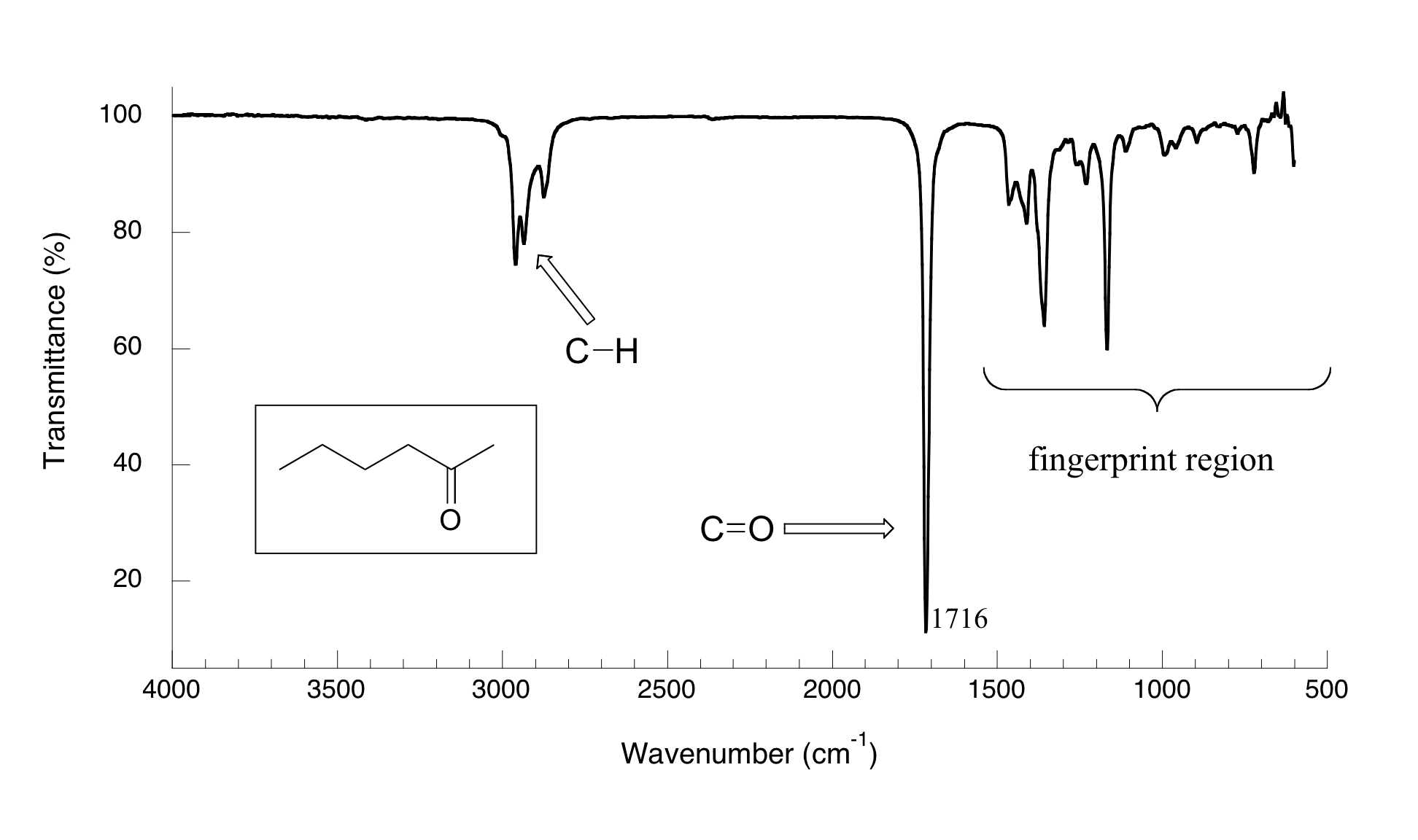
Hay una número de cosas que necesitan ser explicadas con el fin de que usted entienda qué es lo que estamos viendo. En el eje horizontal vemos longitudes de onda IR expresadas en términos de una unidad llamada número de onda (cm-1), que nos dice cuantas ondas caben en un centímetro. En el eje vertical vemos '% de transmisión', que nos dice con qué fuerza la luz fue absorbida en cada frecuencia (100% de transmitancia significa que ninguna absorción ocurrió en esa frecuencia). La línea sólida traza los valores de % de transmisión para cada longitud de onda - los 'picos' (que en realidad apunta hacia abajo) muestran regiones de fuerte absorción. Por alguna razón, es típico en la espectroscopia IR reportar valores de número de onda en lugar de la longitud de onda (en metros) o frecuencia (en Hz). El 'boca abajo' eje vertical, con picos de absorbancia apuntando hacia abajo en lugar de hacia arriba, es también una convención curiosa en la espectroscopia infrarroja. No queremos hacer las cosas demasiado fácil para usted!
El pico de absorción clave en este espectro es el del doble enlace carbonilo, en 1716 cm-1 (correspondiente a una longitud de onda de 5.86 mm, una frecuencia de 5.15 x 1013 Hz, y un valor de ΔE de 4.91 kcal/mol). Note lo fuerte que este pico es, en relación con los otros en el espectro: una fuerte pico en la región de 1650 a 1750 cm-1 es un claro indicativo de la presencia de un grupo carbonilo. Dentro de ese rango, ácidos carboxílicos, ésteres, cetonas y aldehídos tienden a absorber en el extremo menor de longitud de onda (1700-1750-1) mientras que las cetonas y amidas insaturadas conjugadas tienden a absorber en el extremo de longitud de onda mayor (1650-1700 cm-1).
El pico dentado en aproximadamente 2900-3000 cm-1 es característico de los enlaces carbono-hidrógeno tetraédricos. Este pico no es muy útil, ya que casi todas las moléculas orgánicas que tendrás que analizar tienen estos bonos. Sin embargo, puede servir como punto de referencia familiar para orientarse en un espectro.
Usted se dará cuenta de que hay muchos picos adicionales en este espectro en la región de longitud de onda más larga 400 -1400 cm-1. Esta parte del espectro se llama la región de huella dactilar. Aunque por lo general es muy difícil elegir cualquier identificaciones específicas de grupos funcionales de esta región, si contiene información valiosa. La razón de esto es sugerida por el nombre: al igual que una huella digital humana, el patrón de picos de absorbencia en la región de la huella dactilar es única para cada molécula, lo que significa que los datos de una muestra desconocida se puede comparar con los espectros IR de los estándares conocidos con el fin de hacer una identificación positiva. A mediados de los años 1990, por ejemplo, varias pinturas fueron identificadas como falsificaciones porque los científicos pudieron identificar la región de huella de IR de los compuestos de pigmentos rojos y amarillos que no hubieran estado a la disposición del artista que supuestamente creó la pintura (para más detalles ver Chemical and Engineering News, 10 Septiembre, 2007, p. 28).
Ahora, veamos el espectro IR de 1-hexanol.
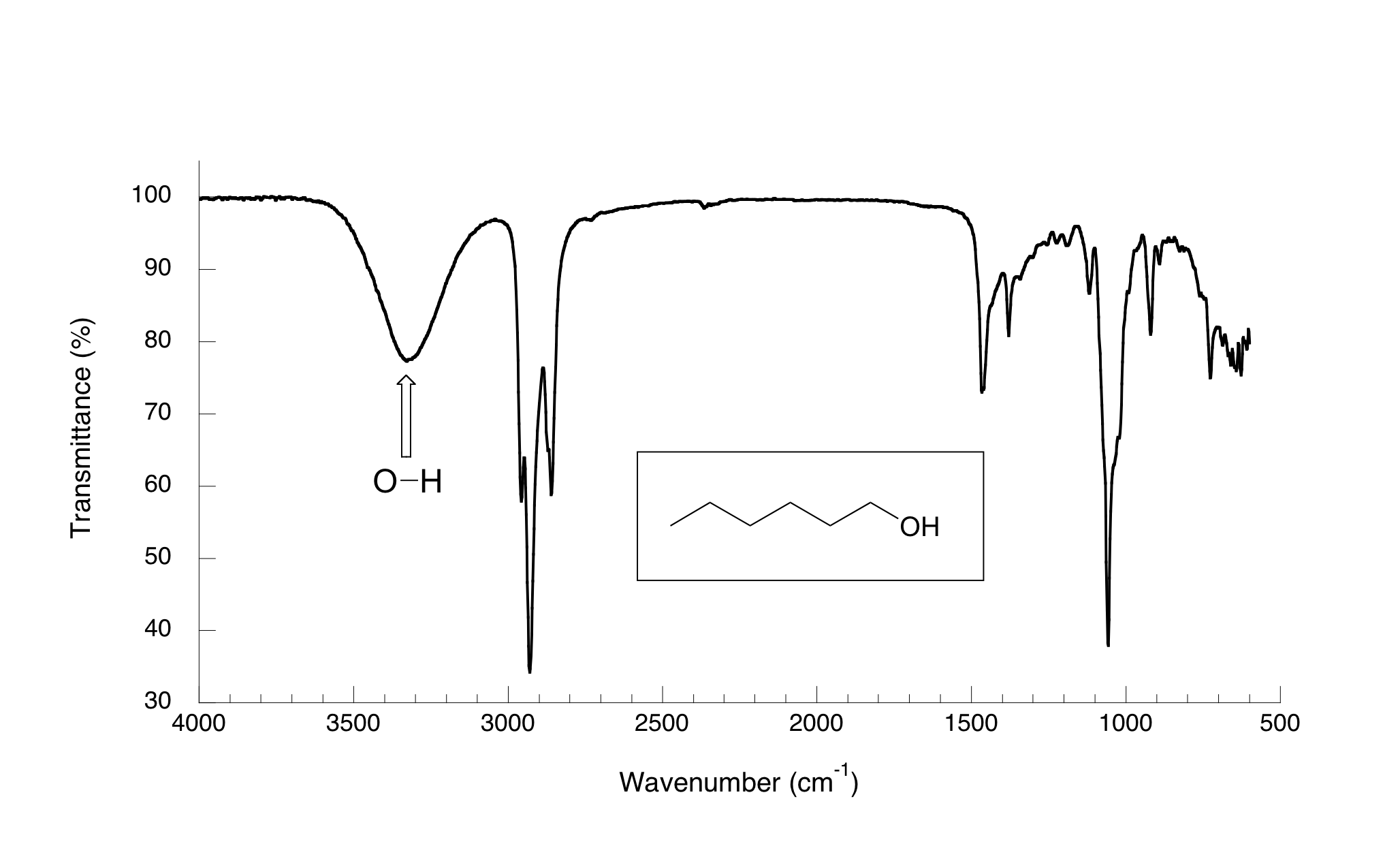
Como puedes ver, el pico de carbonilo ha desaparecido, y en su lugar esta una "montaña" muy amplia centrada alrededor de 3400 cm-1. Esta señal es característica de la forma de estiramiento de O-H en alcoholes, y es un claro indicativo de la presencia de un grupo de alcohol. La amplitud de esta señal es una consecuencia del enlace de hidrógeno entre las moléculas.
En el espectro de ácido octanoico vemos, como se esperaba, el pico característico carbonilo, esta vez en 1709 cm-1.

También vemos una baja, y amplia banda de absorbancia que se ve como un alcohol, excepto que está desplazado ligeramente hacia el lado derecho (longitud de onda larga) del espectro, haciendo que se solapan hasta cierto grado con la región C-H. Esta es la absorbancia de estiramiento característica de ácido carboxílico O-H de enlace simple
El espectro de 1-octeno muestra dos picos que son característicos de alquenos: la de 1642 cm-1 es debido al estiramiento del doble enlace carbono-carbono, y el de 3079 cm-1 se debe al estiramiento de enlace s entre los carbonos de alqueno y sus hidrógenos unidos.
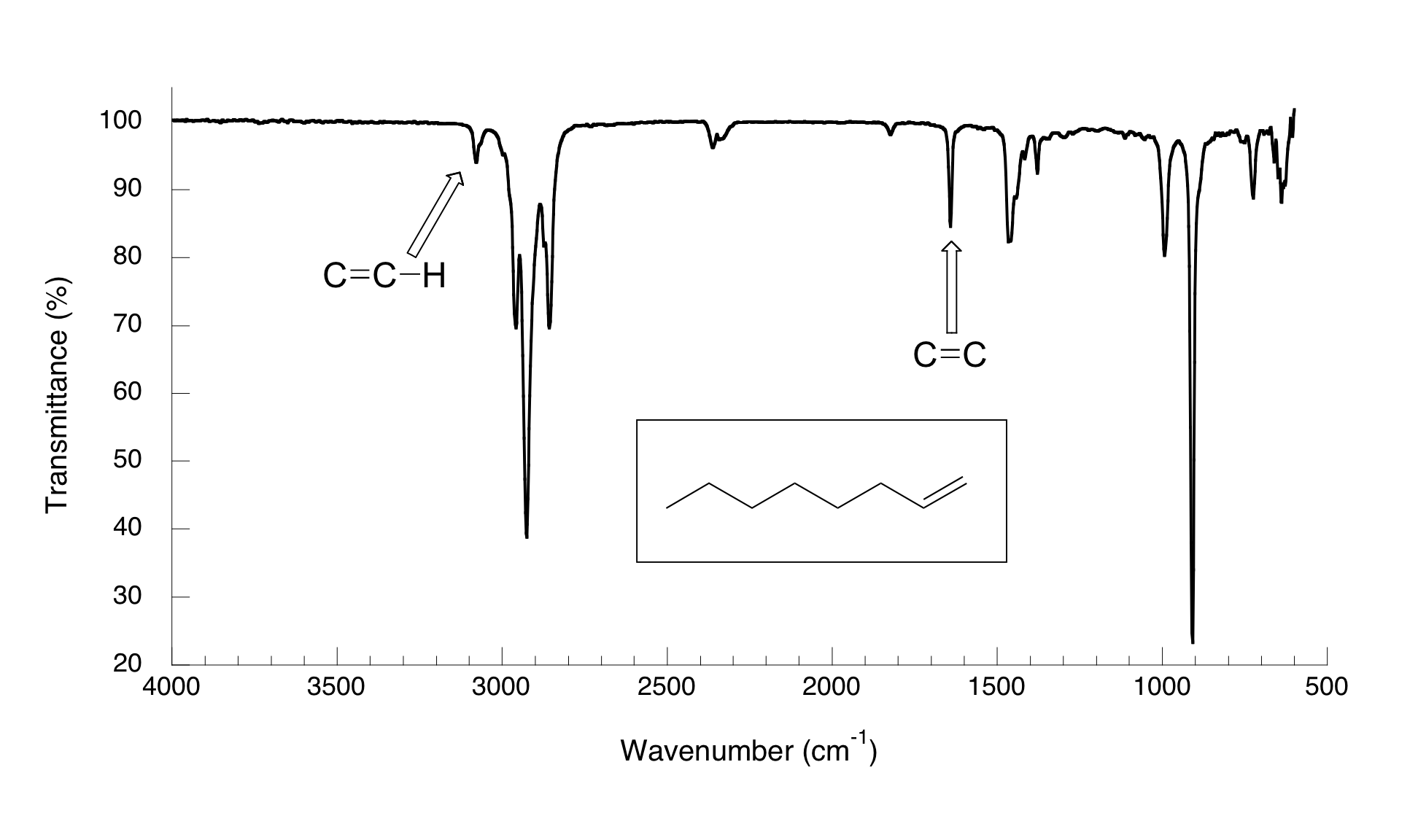
Los alquinos tienen característicos picos de absorbancia IR en el rango de 2100 a 2250 cm-1 debido al estiramiento del triple enlace carbono-carbono, y alquenos terminales pueden ser identificados por su absorbancia a alrededor de 3300 cm-1, debido al estiramiento del enlace entre el carbono sp-hibridado y el hidrógeno terminal.
Es posible identificar otros grupos funcionales tales como aminas y éteres, pero los picos característicos para estos grupos son considerablemente más sutil y / o variable, y a menudo se solapan con los picos de la región de huella dactilar. Por esta razón, limitaremos nuestra discusión a los grupos funcionales más fácilmente reconocibles, cuales se resumen en la tabla 1 en la sección de los cuadros al final del texto.
Como puedes imaginar, obteniendo un espectro de IR para un compuesto no nos permitirá averiguar la estructura completa de incluso una simple molécula, a menos que resulta que tienes un espectro de referencia para la comparación. En conjunción con otros métodos analíticos, sin embargo, espectroscopia IR puede llegar a ser una herramienta muy valiosa, dada la información que proporciona sobre la presencia o ausencia de grupos funcionales claves. IR también puede ser una manera rápida y conveniente para un químico para verificar si una reacción se ha realizado como estaba previsto. Si fuéramos a ejecutar una reacción en la que deseamos convertir ciclohexanona a ciclohexanol, por ejemplo, una rápida comparación de los espectros IR del compuesto inicial y el producto nos dirá si hemos convertido con éxito el grupo cetona a un alcohol (este tipo de reacción se describe en detalle en el capítulo 16).