
6.5: Estructura de las proteínas
- Page ID
- 2356

6.4A: Amino acids and peptide bonds
Proteins are long chains of small ‘building block’ molecules called amino acids. There are twenty different amino acids generally found in proteins, all of which are based on the common structural framework shown below.
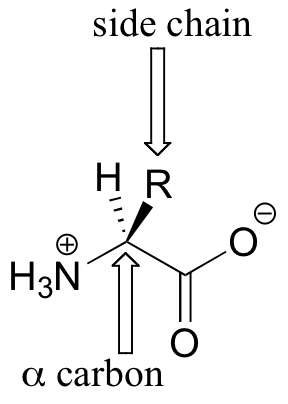
Amino acids contain an amino group on one side and a carboxylate group on the other, with a substituted carbon, referred to as the ‘alpha-carbon’, in the middle. It is the substituent on the alpha-carbon – designated ‘R’ in the general structure here, and often referred to as the amino acid ‘side chain’ – that distinguishes the 20 different amino acids. In all but one of the twenty, the alpha-carbon is a chiral center (in glycine, the ‘R’ group is simply a hydrogen, and thus the alpha-carbon is achiral). Virtually all of the amino acids found in proteins have the stereochemistry shown in the general figure above, and are referred to as L-amino acids ('L' stand for levorotary, the property of rotating plane polarized light in a counter-clockwise direction). Each amino acid is designated by both a three-letter and a single-letter abbreviation: the amino acid alanine, for example, is designated 'Ala' or just 'A'.
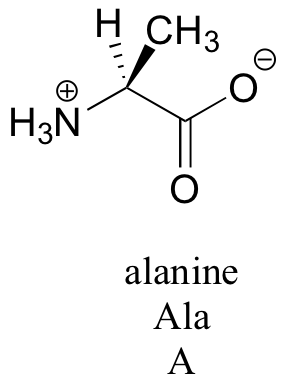
The side chains of amino acids contain a variety of functional groups (see table 5). Some like alanine have only alkyl functionality, while others have alcohol, thiol, sulfide, or amide groups. Four amino acids have aromatic groups on their side chains: phenylalanine has a phenyl group, tyrosine a phenol, tryptophan an indole, and histidine an imidazole. Many of the side chains are ‘ionizable’, capable of acting as acids or bases depending on their protonation state. This property makes them extremely important in enzyme catalysis. Aspartate and glutamate, for example, both have carboxylate groups and are deprotonated at pH 7. Lysine and arginine have amine and imino groups, respectively, and are protonated (and positively charged) at pH 7. Histidine, with its imidazole group, is mainly protonated and positively charged at pH 7. The phenol group of tyrosine is also capable of donating a proton. We will have more to say later about the pKa values of the different side chains in chapter 7. As you progress in your study of organic chemistry, it will become very helpful to familiarize yourself with the structures of all 20 amino acids.
Proteins are formed when amino acids are linked together by ‘peptide bonds’ to form long chains (a peptide bond is really just a specific type of amide functional group). When they are part of a protein chain, individual amino acids are often referred to as 'residues'. The figure below shows a 'dipeptide' - two amino acids linked together by a peptide bond.
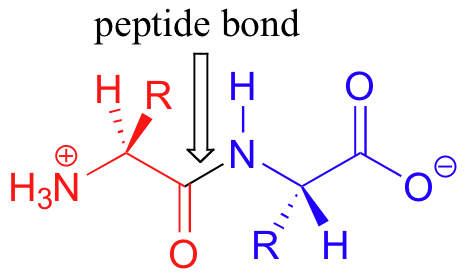
Many protein chains are several hundred amino acids long. The ‘backbone’, or ‘main chain’ of a protein refers to the repeating nitrogen - alpha-carbon – carbonyl pattern: in other words, everything but the side chain. As we have discussed previously, the peptide bond has significant double-bond character due to resonance, and therefore there is a considerable barrier to rotation. Other sigma bonds in proteins, however, have more rotational flexibility.
6.4B: Visualizing protein structure: X-ray crystallography
The key to the amazing versatility of proteins lies in their ability to fold up into very specific three dimensional structures. Much of what we know about the three-dimensional structure of proteins has been gained from a technique called x-ray crystallography. The details are beyond the scope of this text, but the basic idea is fairly easy to grasp. Essentially, patterns formed when x-rays are diffracted by a protein crystal allow for the generation of a three-dimensional picture of electron density. The electron density information can then be translated to the form of a map, on x, y, and z coordinates, of every non-hydrogen atom in the protein. Computer programs can use this data to generate convenient pictures of the protein.
Several of the figures that we’ll be looking at in this section were generated using x-ray crystallography data retrieved from a database called the PDB (Protein Data Bank), and visualized using a program called Jmol. These are free, easy to use web-based resources.
Figures in this section were derived from x-ray crystal structures of the following enzymes:
- fructose 1,6-bisphosphate aldolase
- fructose 6-phosphate aldolase
- DNA adenine methyltransferase
- transaldolase
- protein farnesyltransferase
Click on the links to look at these proteins in Jmol, rotate them around, zoom in and out, and focus on different groups.
6.4C: The four levels of protein structure
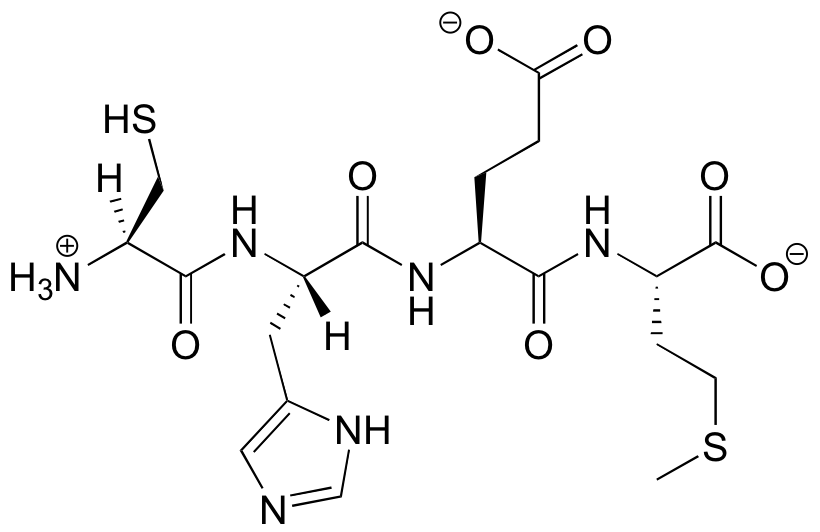
Protein structure can be discussed at four distinct levels. A protein’s primary structure is two-dimensional - simply the sequence of amino acids in the peptide chain.
Below is a Lewis structure of a short segment of a protein with the sequence CHEM (cysteine - histidine - glutamate - methionine)
Secondary structure is three-dimensional, but is a local phenomenon, confined to a relatively short stretch of amino acids. For the most part, there are three important elements of secondary structure: helices, beta-sheets, and loops. In a helix, the main chain of the protein adopts the shape of a clockwise spiral staircase, and the side chains point out laterally.
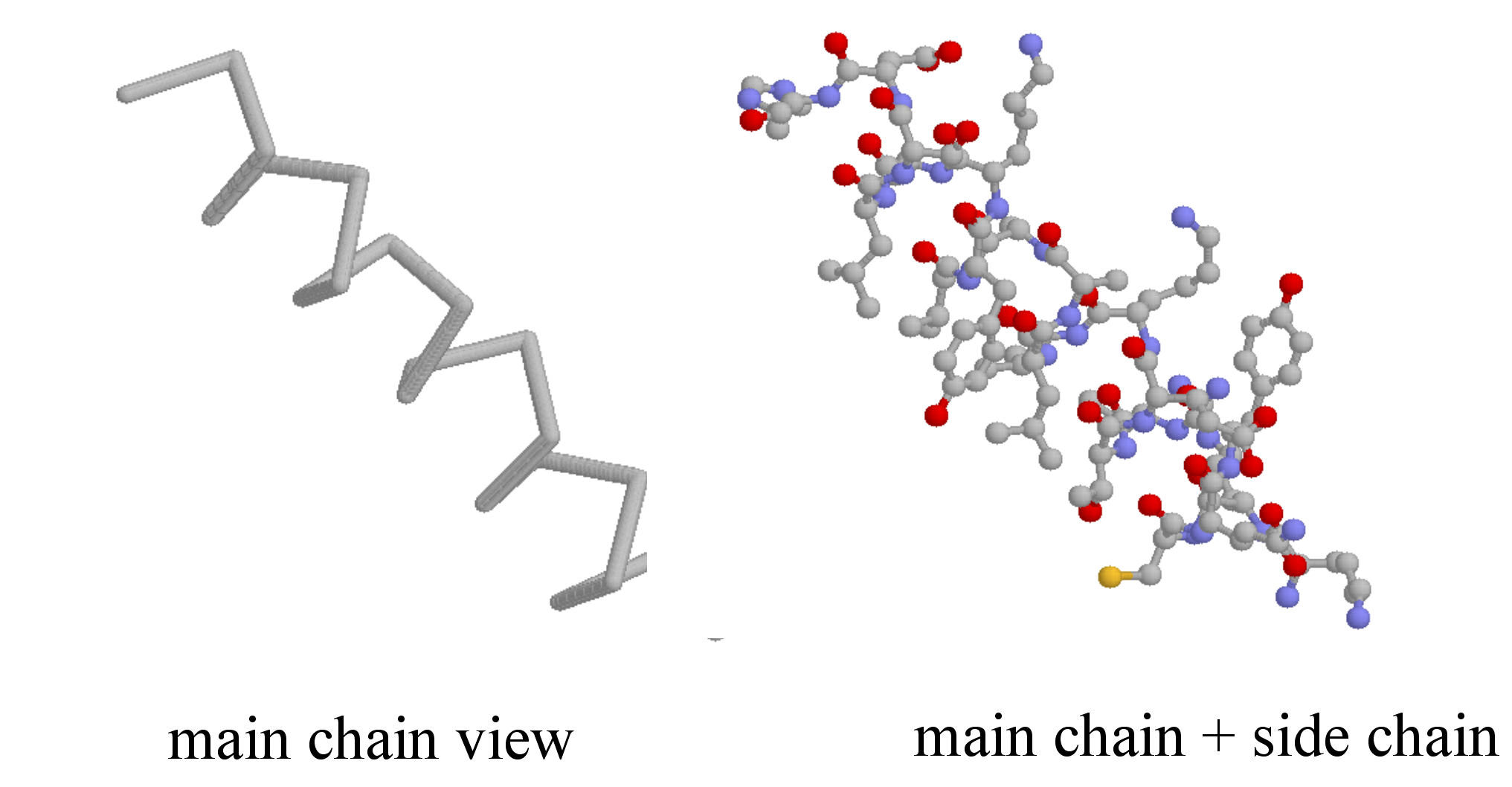
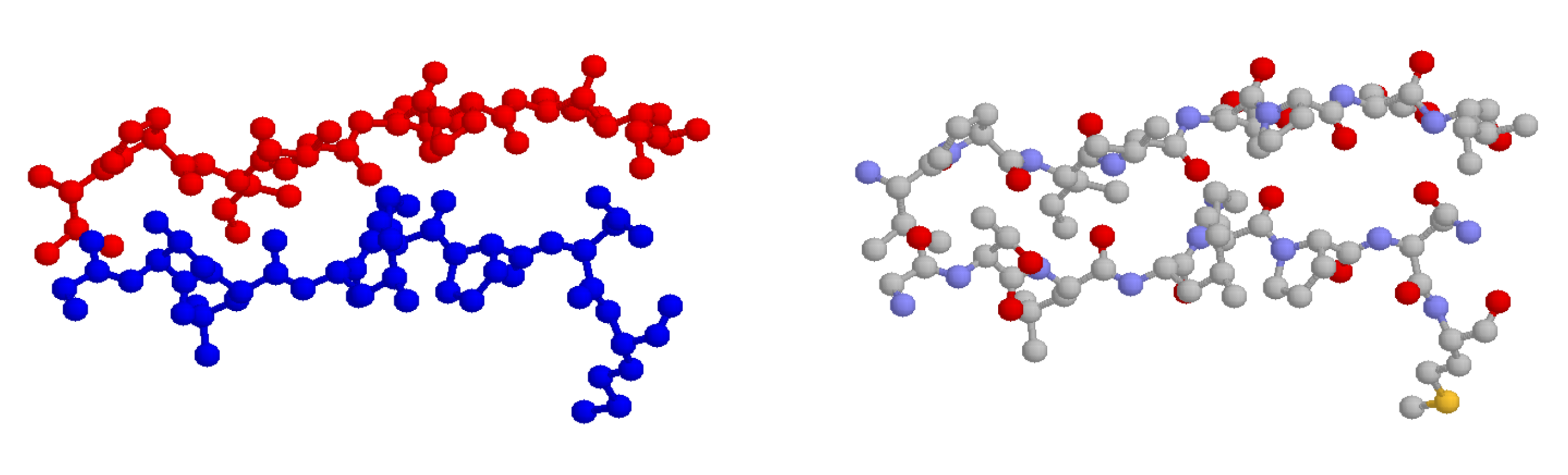
In a beta-sheet (or beta-strand) structure, two sections of protein chain are aligned side-by-side in an extended conformation. The figure below shows two different views of the same beta-sheet: in the left-side view, the two regions of protein chain are differentiated by color.
Loops are relatively disordered segments of protein chain, but often assume a very ordered structure when in contact with a second protein or a smaller organic compound.
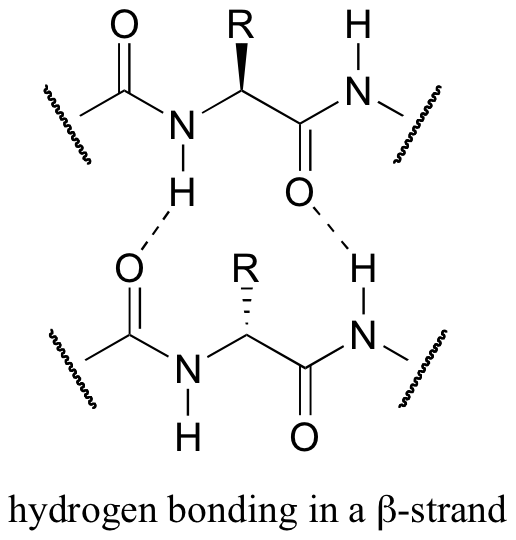
Both helix and the beta-sheet structures are held together by very specific hydrogen-bonding interactions between the amide nitrogen on one amino acid and the carbonyl oxygen on another. The hydrogen bonding pattern in a section of a beta-strand is shown below.
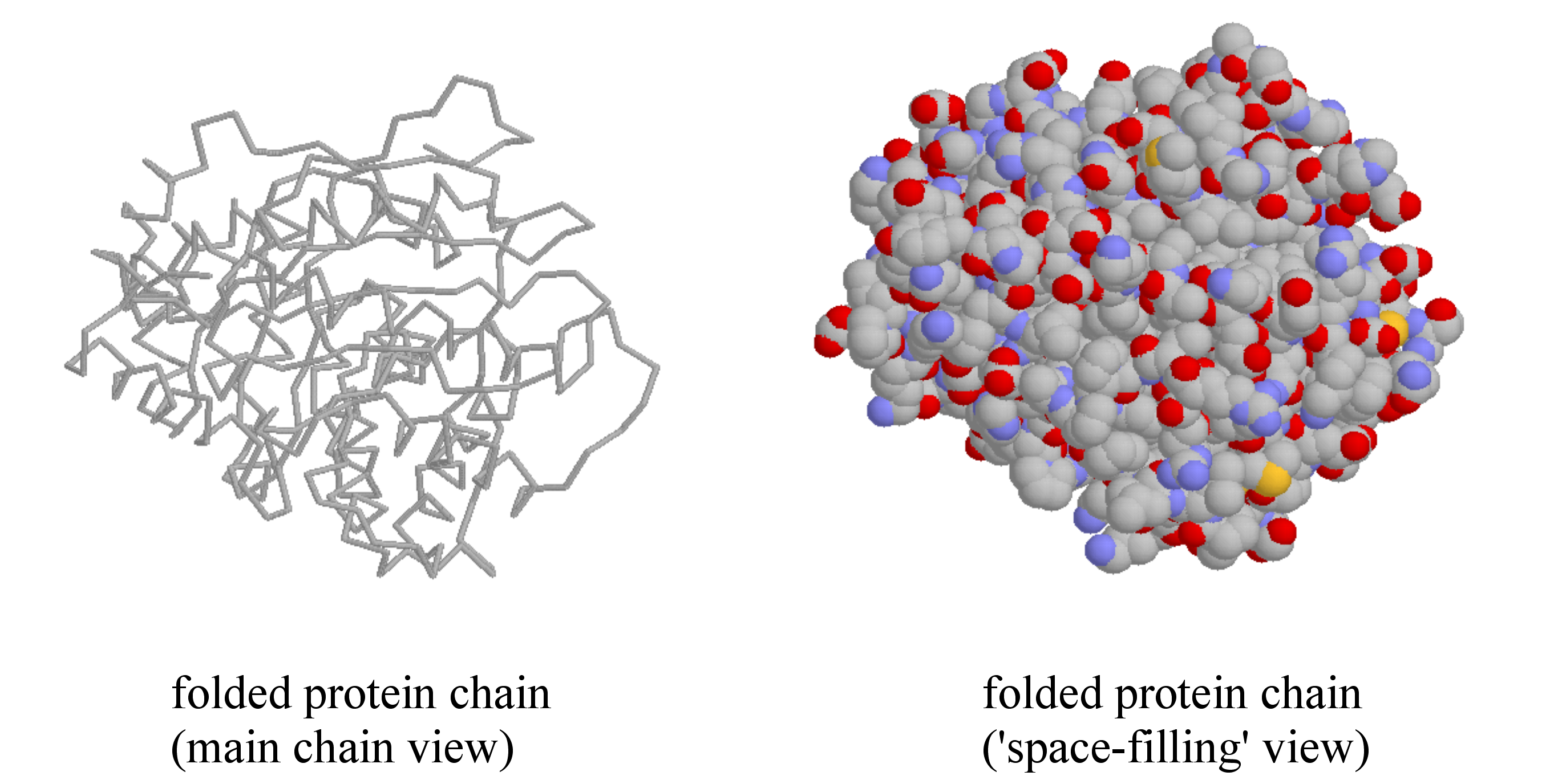
A protein’s tertiary structure is the shape in which the entire protein chain folds together in three-dimensional space, and it is this level of structure that provides protein scientists with the most information about a protein’s specific function.
While a protein's secondary and tertiary structure is defined by how the protein chain folds together, quaternary structure is defined by how two or more folded protein chains come together to form a 'superstructure'. Many proteins consist of only one protein chain, or subunit, and thus have no quaternary structure. Many other proteins consist of two identical subunits (these are called homodimers) or two non-identical subunits (these are called heterodimers).
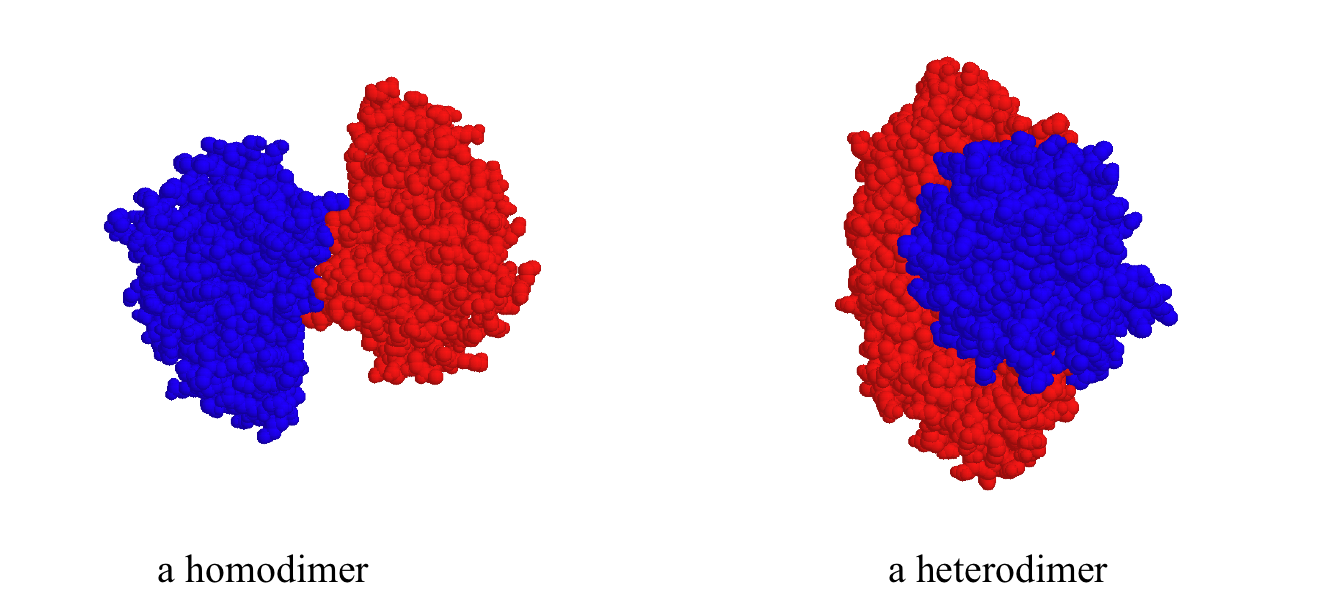
Quaternary structures can be quite elaborate: below we see a protein whose quaternary structure is defined by ten identical subunits arranged in two five-membered rings, forming what can be visualized as a 'double donut' shape (this is fructose 1,6-bisphosphate aldolase):

6.4D: The molecular forces that hold proteins together
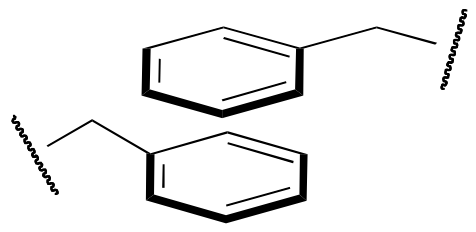
The question of exactly how a protein ‘finds’ its specific folded structure out of the vast number of possible folding patterns is still an active area of research. What is known, however, is that the forces that cause a protein to fold properly and to remain folded are the same basic noncovalent forces that we talked about in chapter 2: ion-ion, ion-dipole, dipole-dipole, hydrogen bonding, and hydrophobic (van der Waals) interactions. One interesting type of hydrophobic interaction is called ‘aromatic stacking’, and occurs when two or more planar aromatic rings on the side chains of phenylalanine, tryptophan, or tyrosine stack together like plates, thus maximizing surface area contact.
Hydrogen bonding networks are extensive within proteins, with both side chain and main chain atoms participating. Ionic interactions often play a role in protein structure, especially on the protein surface, as negatively charged residues such as aspartate interact with positively-charged groups on lysine or arginine.
One of the most important ideas to understand regarding tertiary structure is that a protein, when properly folded, is polar on the surface and nonpolar in the interior. It is the protein's surface that is in contact with water, and therefore the surface must be hydrophilic in order for the whole structure to be soluble. If you examine a three dimensional protein structure you will see many charged side chains (e.g. lysine, arginine, aspartate, glutamate) and hydrogen-bonding side chains (e.g. serine, threonine, glutamine, asparagine) exposed on the surface, in direct contact with water. Inside the protein, out of contact with the surrounding water, there tend to be many more hydrophobic residues such as alanine, valine, phenylalanine, etc. If a protein chain is caused to come unfolded (through exposure to heat, for example, or extremes of pH), it will usually lose its solubility and form solid precipitates, as the hydrophobic residues from the interior come into contact with water. You can see this phenomenon for yourself if you pour a little bit of vinegar (acetic acid) into milk. The solid clumps that form in the milk are proteins that have come unfolded due to the sudden acidification, and precipitated out of solution.
In recent years, scientists have become increasing interested in the proteins of so-called ‘thermophilic’ (heat-loving) microorganisms that thrive in hot water environments such as geothermal hot springs. While the proteins in most organisms (including humans) will rapidly unfold and precipitate out of solution when put in hot water, the proteins of thermophilic microbes remain completely stable, sometimes even in water that is just below the boiling point. In fact, these proteins typically only gain full biological activity when in appropriately hot water - at room-temperature they act is if they are ‘frozen’. Is the chemical structure of these thermostable proteins somehow unique and exotic? As it turns out, the answer to this question is ‘no’: the overall three-dimensional structures of thermostable proteins look very much like those of ‘normal’ proteins. The critical difference seems to be simply that thermostable proteins have more extensive networks of noncovalent interactions, particularly ion-ion interactions on their surface, that provides them with a greater stability to heat. Interestingly, the proteins of ‘psychrophilic’ (cold-loving) microbes isolated from pockets of water in arctic ice show the opposite characteristic: they have far fewer ion-ion interactions, which gives them greater flexibility in cold temperatures but leads to their rapid unfolding in room temperature water.