
15.3: Isomerización y sustitución electrofílica (adición-eliminación)
- Page ID
- 2429

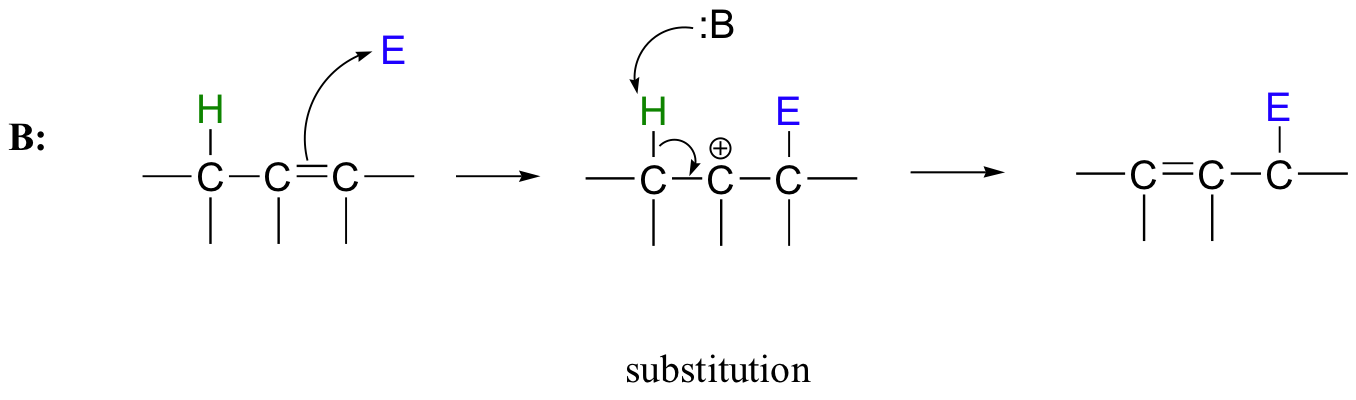
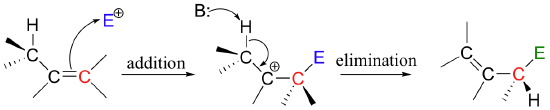
Many electrophilic reactions do not result in the conversion of an alkene to a substituted alkane, as is the case with electrophilic additions. In many cases, attack by the π bond is followed by elimination, which re-establishes the carbon-carbon double bond. This can result either in a change in location of the double bond (an isomerization - pathway A below), or substitution of an electrophile for a proton (pathway B below)
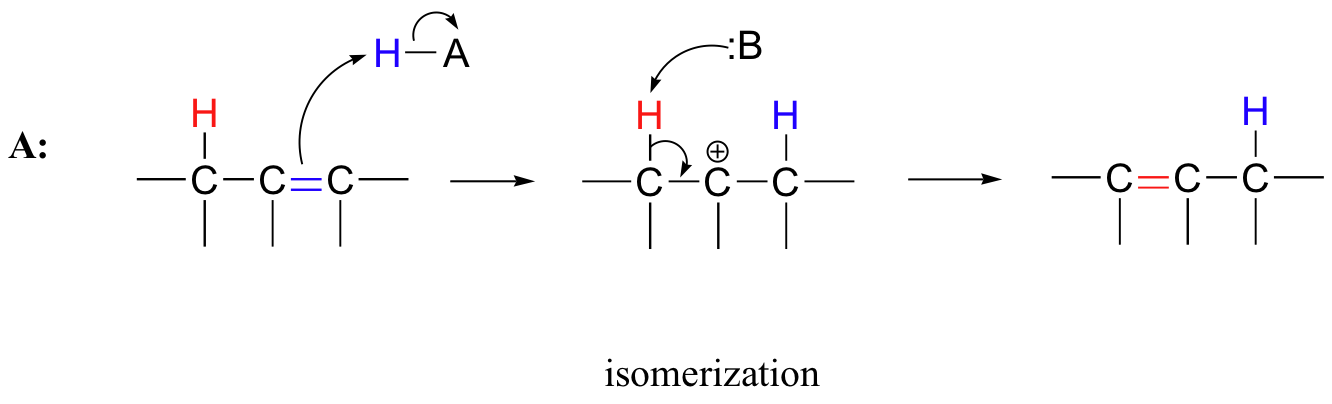
15.3A: Alkene isomerization
Recall from section 13.2C that the position of an alkene double bond is 'shuffled' (isomerized) in the breakdown of unsaturated fatty acids. That reaction proceeded through a negatively-charged, enolate intermediate. Alkenes can also undergo positional isomerization through an electrophilic, carbocation intermediate mechanism in molecules with no adjacent carbonyl group. In this case, protonation of the substrate occurs first, followed by deprotonization (the opposite order of the enolate mechanism).
In the process of isoprenoid chain construction, isopentenyl diphosphate (IPP), which is the essential 'building block' for all isoprenoid molecules, is first isomerized to dimethylallyl diphosphate (DMAPP) by an enzyme called 'IPP isomerase'.
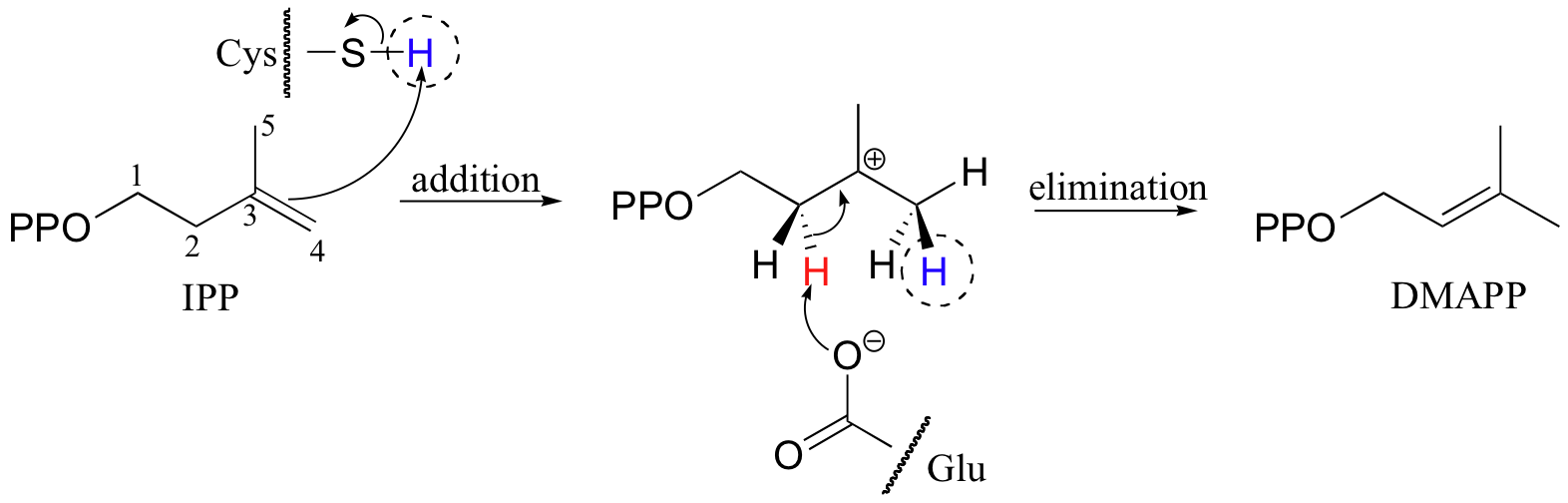
In the first step of the isomerization reaction, the π electrons abstract a proton from an active site cysteine side chain. In an electrophilic addition reaction, a nucleophile such as water would then quench the carbocation, forming a tertiary alcohol. In the isomerase reaction, however, x-ray crystallography studies (EMBO J. 2001, 20, 1530) show that the intermediate is bound in a very deep active site cavity in order to protect it from just such an attack by solvent water. Instead, a glutamate residue acts as a base, abstracting a proton from C2 of the intermediate to initiate an elimination. The crystal structure shows that the acid/base pair in the active site - the cysteine and the glutamate - are located on opposite sides of the active site, implying that a proton is added on one side of the molecule and removed from the other. This is referred to as an antarafacial transposition of a proton. If protonation and subsequent deprotonation occur on the same side of a molecule, it is referred to as a suprafacial transposition.
15.3B: Substitution by electrophilic addition/elimination
The electrophilic double bond isomerization catalyzed by IPP isomerase is a highly reversible reaction, with an equilibrium IPP:DMAPP ratio of about 6:1. In the next step of isoprenoid biosynthesis, the two five-carbon isomers condense to form a 10-carbon isoprenoid product called geranyl diphosphate (GPP).
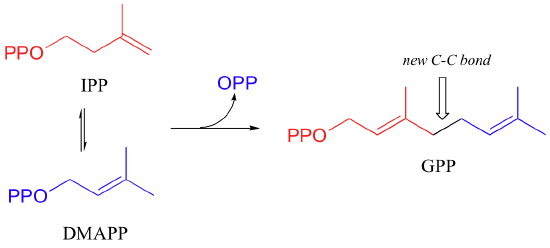
This is a nice example of an electrophilic addition/elimination mechanism, which we saw in general form in section 15.1:
The first step is ionization of the electrophile - in other words, the leaving group departs and a carbocation intermediate is formed. In this case, the pyrophosphate group on DMAPP is the leaving group, and the electrophilic species is the resulting allylic carbocation.
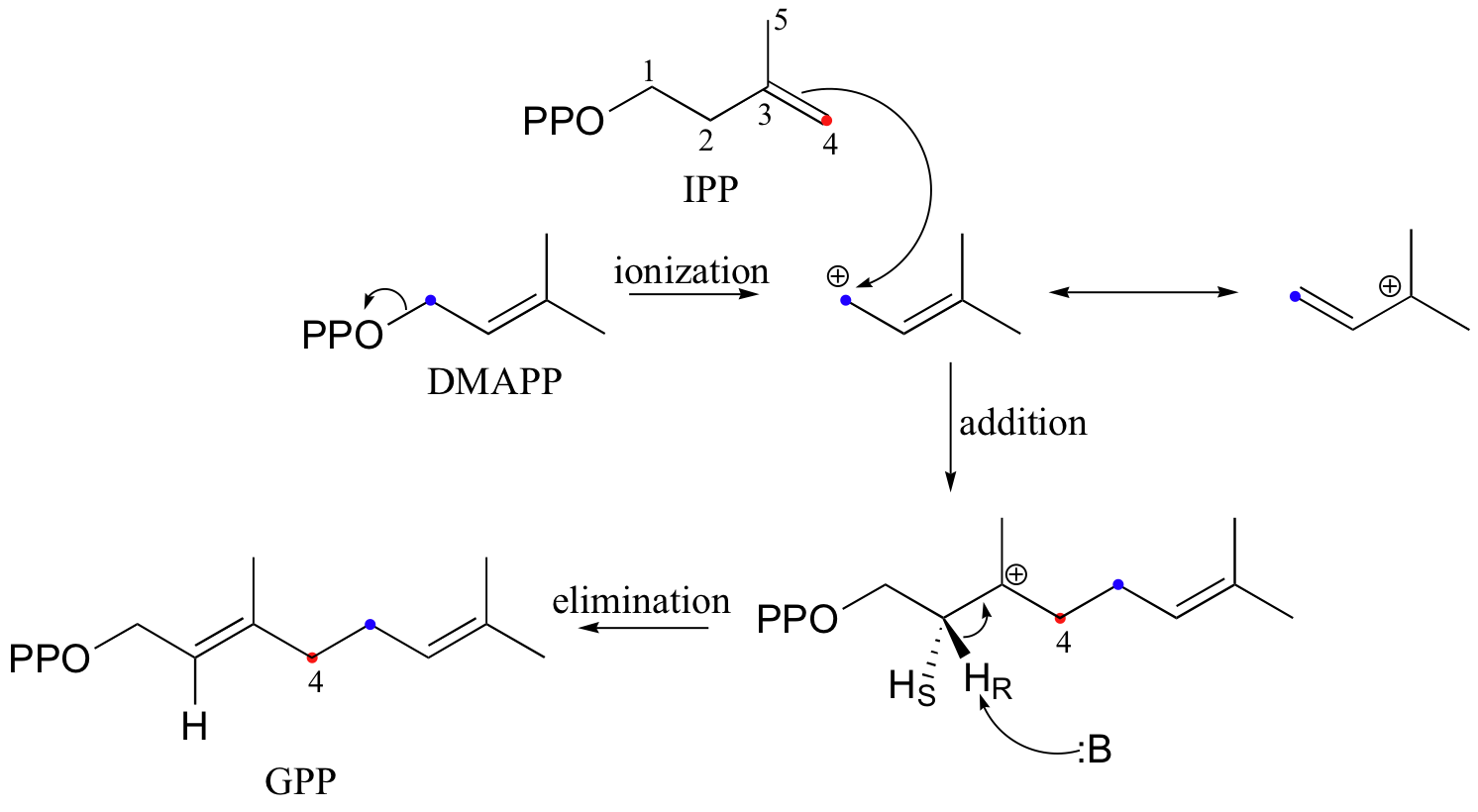
In the condensation (addition) step, the C3-C4 double bond in IPP attacks the positively-charged C1 of DMAPP, resulting in a new carbon-carbon bond and a second carbocation intermediate, this time at a tertiary carbon. In the elimination phase, proton abstraction leads to re-establishment of a double bond in the GPP product. Notice that the enzyme specifically takes the pro-R proton in this step.
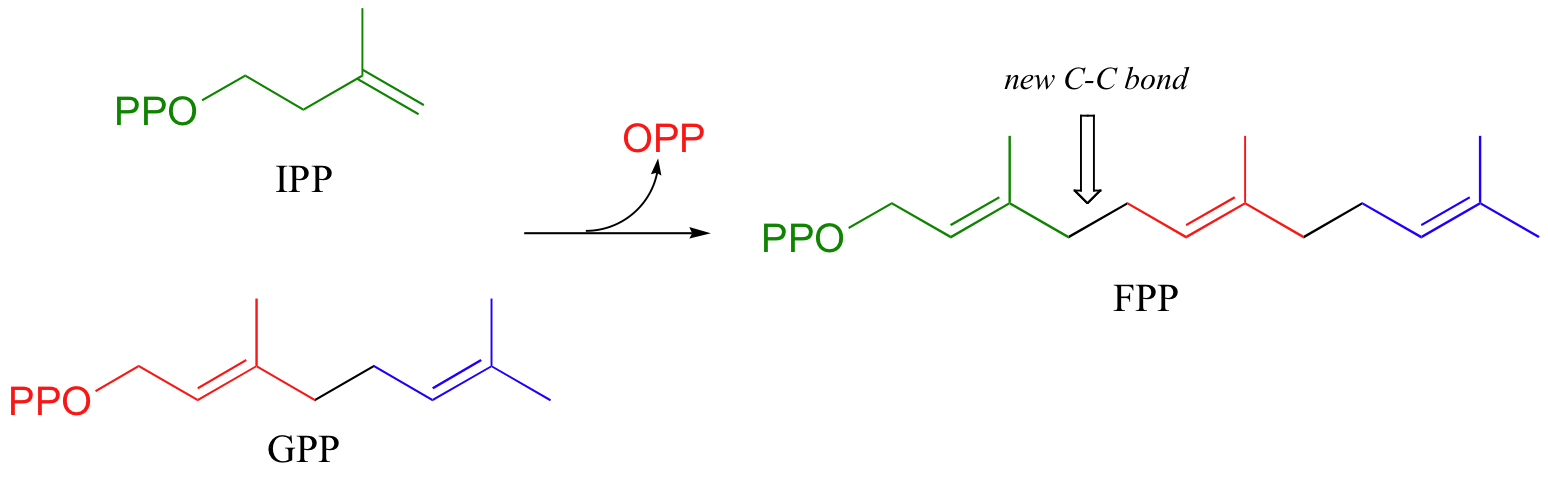
To continue the chain elongation process, another IPP molecule can then condense, in a very similar reaction, with C1 of geranyl diphosphate to form a 15-carbon product called farnesyl diphosphate (FPP).
How do we know that these are indeed SN1-like mechanisms with carbocation intermediates, rather than concerted SN2-like mechanisms? First of all, recall that the question of whether a substitution is dissociative (SN1-like) or associative (SN2-like) is not always clear-cut - it could be somewhere in between, like the protein prenyltransferase reaction (section 9.3). The protein prenyltransferase reaction and the isoprenoid chain elongation reactions are very similar: the electrophile is the same, but in the former the nucleophile is a thiolate, while in the latter the nucleophile is a pi bond.
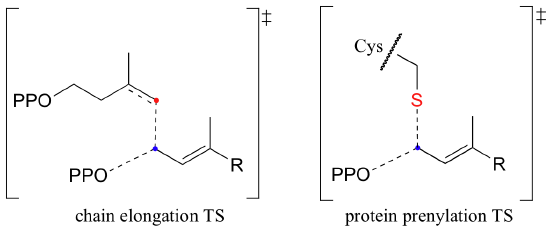
This difference in the identity of the nucleophilic species would lead one to predict that the chain elongation reaction has more SN1-like character than the protein prenylation reaction. A thiolate is a very powerful nucleophile, and thus is able to push the pyrophosphate leaving group off, implying some degree of SN2 character. The electrons in a pi bond, in contrast, are only weakly nucleophilic, and thus need to be pulled in by a powerful electrophile - ie. a carbocation.
So it makes perfect sense that the chain elongation reaction should more SN1-like than SN2-like. Is this in fact the case? We know how to answer this question experimentally - just run the reaction with fluorinated DMAPP or GPP substrates and observe how much the fluorines slow things down (see section 9.3B).
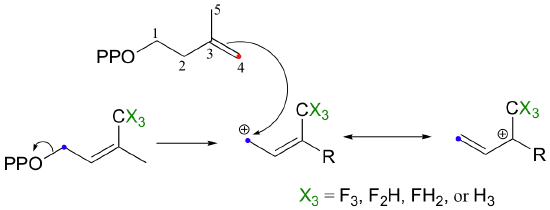
If the reaction is SN1-like, the electron-withdrawing fluorines should destabilize the allylic carbocation intermediate and thus slow the reaction down considerably. If the mechanism is SN2-like, the fluorine substitutions should not have a noticeable effect, because a carbocation intermediate would not be formed. When this experiment was performed with FPP synthase, the results were dramatic: the presence of a single fluorine slowed down the rate of the reaction by a factor of about 60, while two and three fluorines resulted in a reaction that was 500,000 and 3 million times slower, respectively (J. Am. Chem. Soc. 1981, 103, 3926.) These results strongly suggest indicate the formation of a carbocation intermediate in an SN1-like displacement.
Template:Soderberg