
14.2: Variaciones en la reacción Michael
- Page ID
- 2420

14.2A: cis/trans alkene isomerization
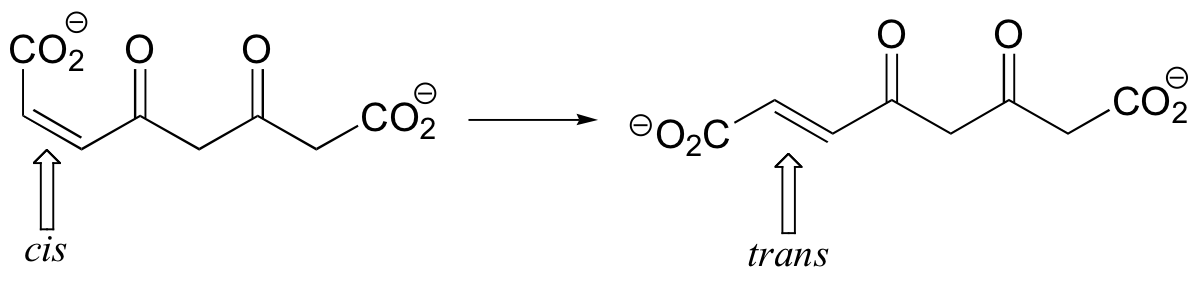
Some enzymes are able to catalyze the stereoisomerization of alkene groups using a Michael addition step. Maleylacetoacetate isomerase catalyzes the following cis to trans alkene isomerization as part of the degradation of the aromatic amino acids phenylalanine and tyrosine.
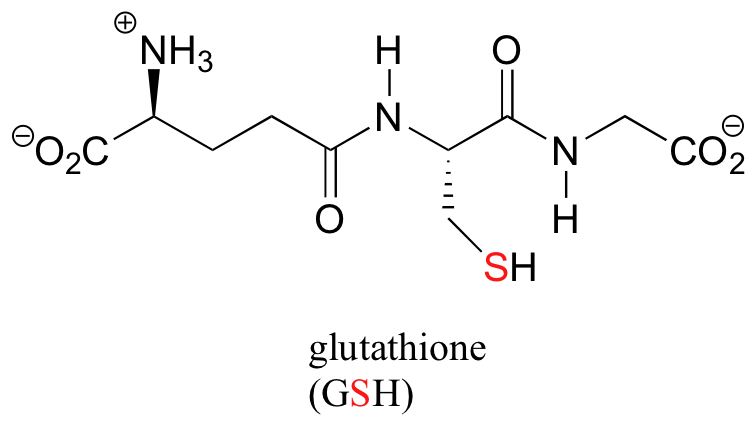
An important player in this reaction is a tripeptide coenzyme called glutathione, commonly abbreviated GSH. The important part of glutathione in terms of its chemical activity is the nucleophilic cysteine thiol group - hence the abbreviation GSH.
In effect, GSH is just acting as a 'nucleophile for hire' in the maleylacetoacetate isomerase reaction. In the first step, it attacks the beta-carbon in a Michael addition.
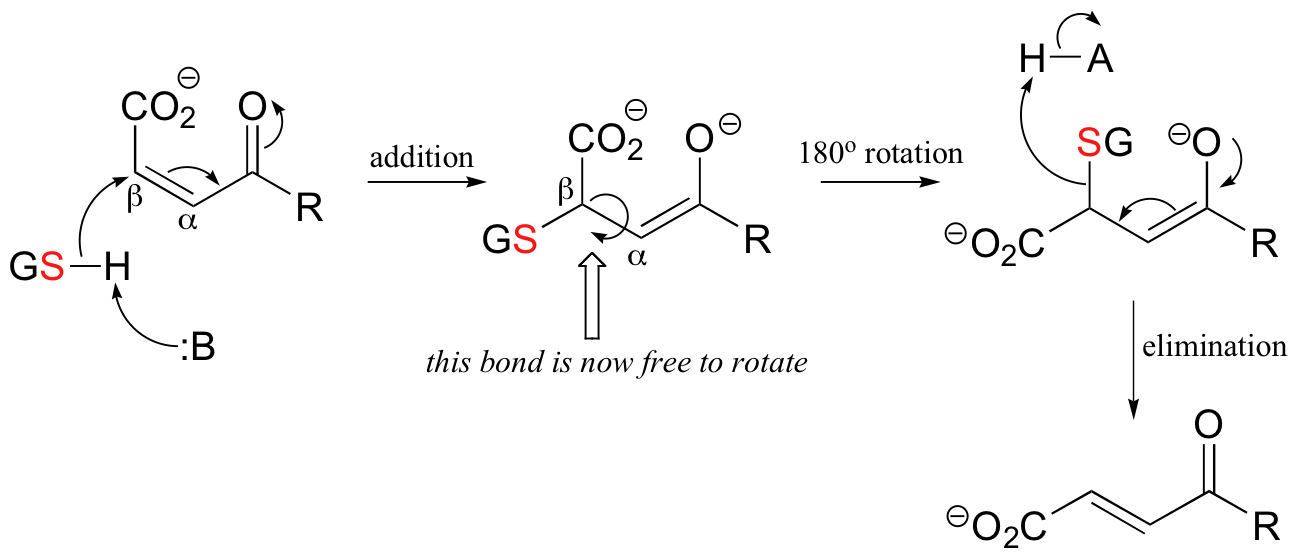
Notice what this accomplishes: the Cα-Cβ linkage no longer involves a pi-bond (because Cβ is now sp3-hybridized!) and thus is free to rotate - and it does, by 180o. After rotation is complete, the Michael enolate simply collapses, reforming the Cα-Cβ pi-bond in the trans configuration and eliminating GSH.
14.2B: Nucleophilic aromatic substitution
Sometimes, a reaction that starts out with a conjugate addition step ends up as a substitution rather than an addition. These reactions are most common on aromatic rings. A nucleophilic aromatic substitution reaction starts out with a nucleophilic attack on a carbon electrophile that is conjugated to an electron-withdrawing groups such as a carbonyl or (more likely in laboratory reactions) a nitro group, forming an enolate-like intermediate.

In a Michael addition, the next step would be protonation. However, this is not what happens in this case!
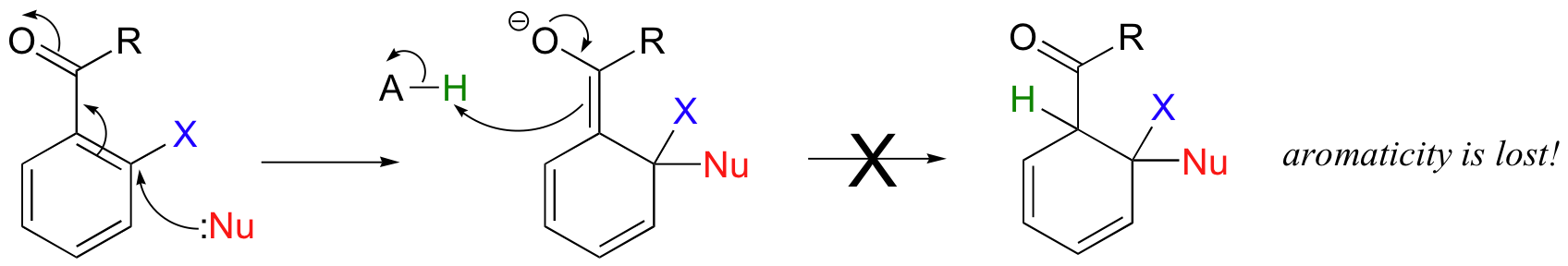
Nucleophilic aromatic substitutions proceed in a completely different direction due to two factors. First, protonation as in a Michael addition would lead to a non-aromatic product, which would be energetically unfavorable (remember that aromatic systems confer a extra degree of stability). Second, substrates in aromatic substitutions have a leaving group (designated 'X' in this general mechanism) at the original site of nucleophilic attack. Instead of abstracting a proton from the solvent, therefore, the reactive electrons return to the aromatic system, simultaneously expelling the leaving group.
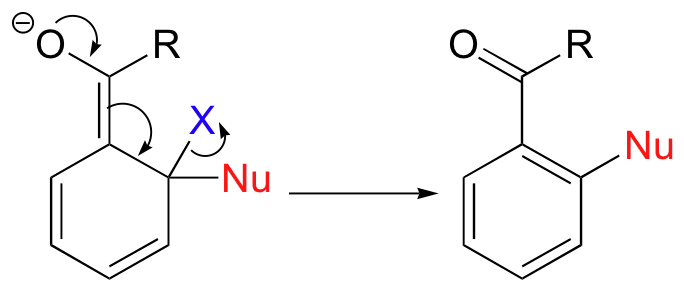
Nucleophilic aromatic substitution can take place at the para position as well as the ortho position, but not at the meta position.
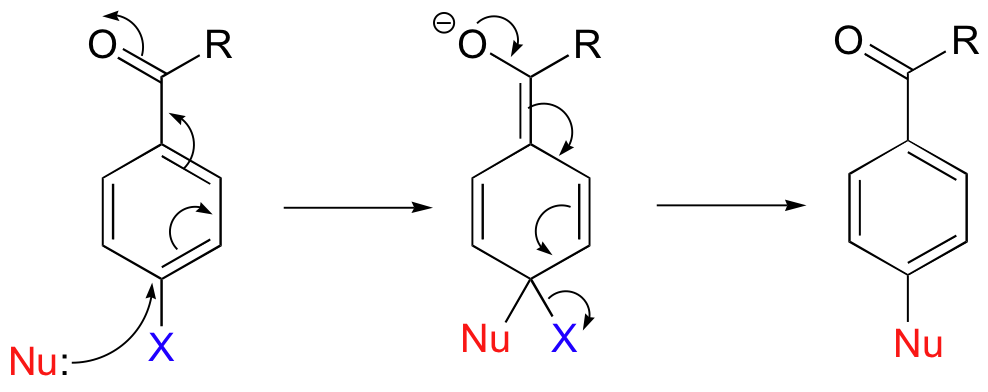
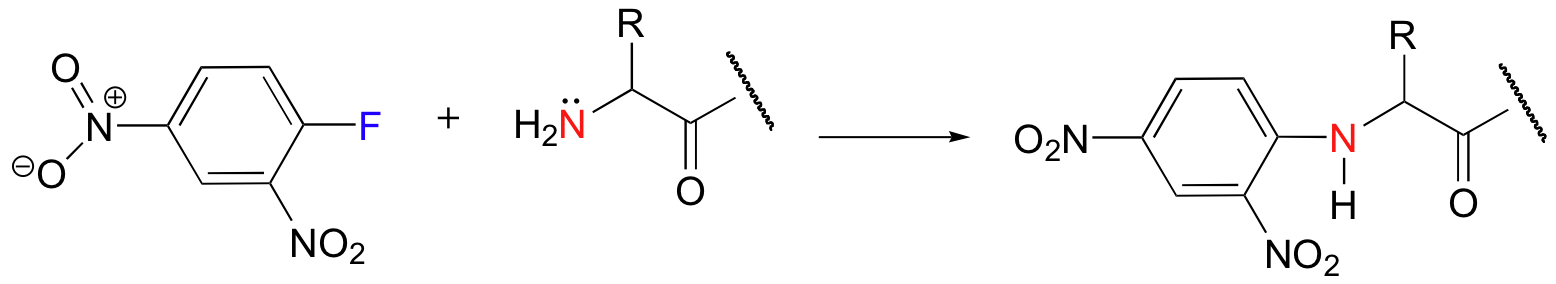
This reaction was developed by Frederick Sanger, a pioneer in protein chemistry.
a) Draw a complete mechanism for the reaction above, clearly showing how the nitro groups stabilize the negative charge on the intermediate.
b) Use a resonance argument to explain how the presence of a second nitro group serves to further activate the aromatic ring for nucleophilic aromatic substitution.
c) Why do you think a flourine substituent is used on the aromatic ring instead of a better leaving group such as bromine or chlorine?

The first step is nucleophilic aromatic substitution, the second is a tautomerization.
14.2C: Synthetic parallel - Michael addition reactions in the laboratory
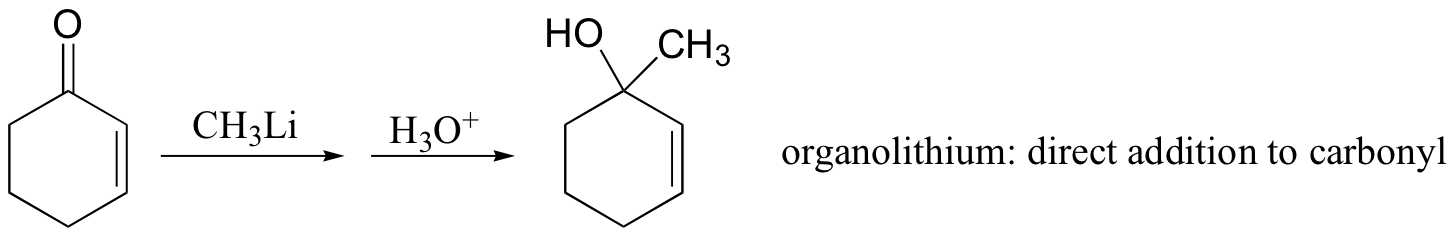
Michael addition reactions are also common in the organic synthesis lab. Interestingly, while organolithium reagents (section 13.6D) react with α,β-unsaturated ketones mainly with 1,2 regiochemistry (ie. attack at the carbonyl), Gilman (lithium diorganocopper) reagents add mainly to the beta-carbon. Grignard reagents usually give a mixture of both regiochemical outcomes.
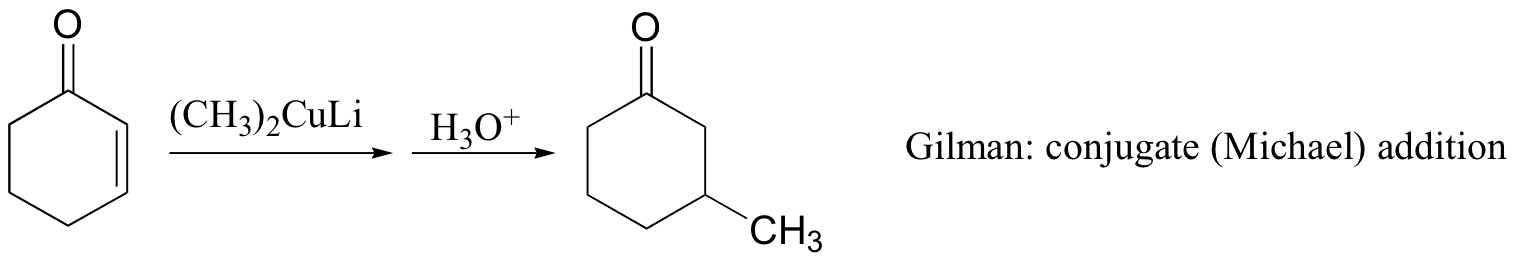
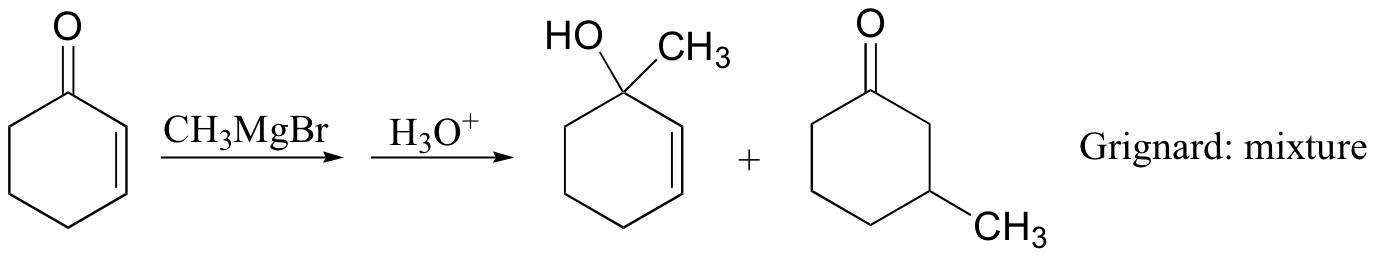
The Stork enamine (section 13.6A) is also frequently used as a nucleophile in carbon-carbon bond-forming Micheal addition reactions in organic synthesis.