
8.3: Hidruros
- Page ID
- 69470

Trihidruros del Grupo 15 Elementos
Los cinco elementos del Grupo 15 forman hidruros de fórmula EH 3. La tabla\(\PageIndex{1}\) enumera los nombres de la IUPAC junto con los de uso más común.
| Compuesto | Nombre tradicional | Nombre de la IUPAC |
|---|---|---|
| NH 3 | Amoníaco | Azane |
| PH 3 | Fosfina | Fosfano |
| aSH 3 | Arsina | Arsane |
| SbH 3 | Stibine | Stibane |
| BiH 3 | bismutina | bismuto |
El punto de ebullición y el punto de fusión aumentan al bajar el Grupo (Cuadro\(\PageIndex{2}\)) con mayor masa molecular, a excepción del NH 3 cuyos puntos de fusión y ebullición anómalamente altos (Figura\(\PageIndex{1}\)) son consecuencia de fuertes N-H... H enlace de hidrógeno. Se observa un efecto similar (y más fuerte) para los hidruros del Grupo 16 (H 2 E).
| Compuesto | Mp (°C) | Bp (°C) | ΔH f (kJ/mol) | Energía de enlace E-H (kJ/mol) | Ángulo de unión H-E-H (°) |
|---|---|---|---|---|---|
| NH 3 | -77.7 | -33.35 | -46.2 | 391 | 107 |
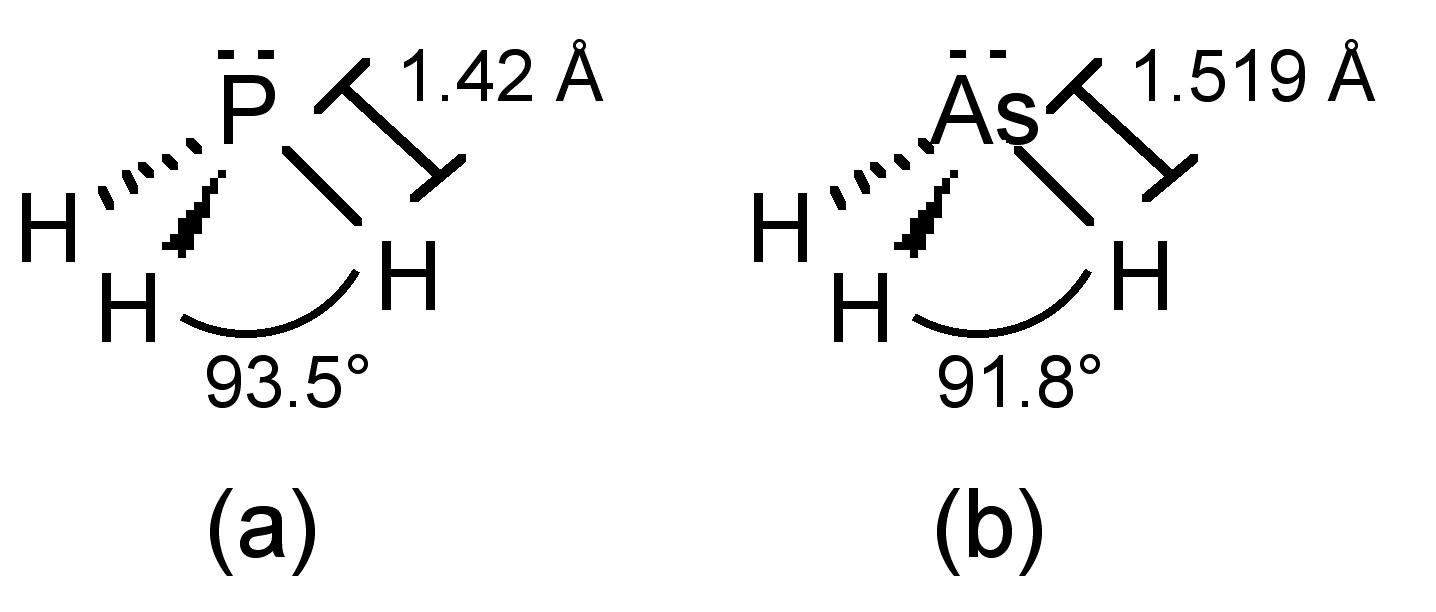
| PH 3 | -133 | -87.7 | 9.3 | 322 | 93.5 |
| aSH 3 | -116.3 | -55 | 172.2 | 247 | 92 |
| SbH 3 | -88 | -17.1 | 142.8 | 255 | 91.5 |

Las fuerzas de unión E-H disminuyen en el grupo y esto se correlaciona con la estabilidad general de cada compuesto (Tabla\(\PageIndex{2}\)). Los ángulos de enlace H-E-H (Tabla\(\PageIndex{2}\)) también disminuyen en el Grupo. Se espera que el ángulo de unión H-E-H sea un ideal tetraédrico de 109.5°, pero dado que los pares solitarios repelen más que los pares de unión, se esperaría que el ángulo real fuera ligeramente menor. Dos posibles explicaciones son posibles para la diferencia entre NH 3 y los otros hidruros.
- El enlace N-H es corto (1.015 Å) en comparación con los análogos más pesados, y el nitrógeno es más electronegativo que el hidrógeno, por lo que el par de enlaces residirá más cerca del átomo central y los pares de enlaces se repelerán entre sí abriendo el ángulo H-N-H más de lo observado para PH 3, etc.
- La accesibilidad de los orbitales 2 s y 2 p sobre nitrógeno permite la hibridación y los orbitales asociados con la unión N-H en NH 3 son, por lo tanto, cercanos a sp 3 en carácter, dando como resultado una geometría cercana a tetraédrica. En contraste, la hibridación de los orbitales n s y n p para P, As, etc., es menos accesible, y como consecuencia los orbitales asociados con la unión P-H en PH 3 están más cerca de p en carácter dando como resultado un ángulo H-P-H de casi 90°. Cuanto más abajo del Grupo es el átomo central menos hibridación se produce y más cerca del carácter p puro los orbitales en E asociados con el enlace E-H.
Amoníaco
El amoníaco (NH 3) es un gas incoloro y acre (bp = -33.5 °C) cuyo olor puede detectarse en concentraciones tan bajas de 20 — 50 ppm. Su alto punto de ebullición en relación con sus congéneres más pesados es indicativo de la formación de fuertes enlaces de hidrógeno. El fuerte enlace de hidrógeno también da como resultado un alto calor de vaporización (23.35 kJ/mol) y, por lo tanto, el amoníaco se puede usar convenientemente como líquido a temperatura ambiente a pesar de su bajo punto de ebullición.
ADVERTENCIA
La solución de amoníaco causa quemaduras e irritación en los ojos y la piel. El vapor causa irritación severa en el sistema respiratorio. Si se ingiere la solución causa graves daños internos.
Síntesis
El amoníaco se fabrica a escala industrial mediante el proceso Haber utilizando la reacción directa de nitrógeno con hidrógeno a alta presión (10 2 — 10 3 atm) y alta temperatura (400 — 550 °C) sobre un catalizador (e.g., α-hierro), (8.3.1).
\[\text{N}_2\text{ + 3 H}_2\rightarrow \text{2 NH}_3\]
En menor escala, el amoníaco se prepara mediante la reacción de una sal de amonio con una base, (8.3.2), o hidrólisis de un nitruro, (8.3.3). Este último es una ruta conveniente a ND 3 mediante el uso de D 2 O.
\[\text{NH}_4\text{X + OH}^- \rightarrow \text{NH}_3\text{ + H}_2\text{O + X}^-\]
\[\text{Mg}_3\text{N}_2\text{ + H}_2\text{O} \rightarrow \text{3 Mg(OH)}_2 \text{ + 2 NH}_3\]
Estructura
El nitrógeno en amoníaco adopta hibridación sp 3, y el amoníaco tiene una estructura paraguas (Figura\(\PageIndex{2}\)) debido al par solitario estereoquímicamente activo.
La barrera a la inversión del paraguas es muy baja (E a = 24 kJ/mol) y la inversión ocurre 100 veces por segundo. Como consecuencia, no es posible aislar aminas quirales de la misma manera que es posible para las fosfinas.
De manera similar al agua, (8.3.4), el amoníaco es un autoionizante, (8.3.5); sin embargo, la constante de equilibrio (K = 10-33) es mucho menor que el agua (K = 10-14). La menor constante dieléctrica del amoníaco (16.5 @ 20 °C) en comparación con el agua (80.4 @ 20 °C) significa que el amoníaco no es tan bueno como el agua como solvente para compuestos iónicos, pero es mejor para compuestos orgánicos covalentes.
\[\text{2 H}_2\text{O} \rightleftharpoons \text{H}_3\text{O}^+\text{ + OH}^-\]
\[\text{2 NH}_3 \rightleftharpoons \underset{\text{ammonium}}{\textext{NH}_4^+} \t{ + } \und\terset{ext{amide}}{\text{NH}_2^- }\]
Reacciones
La similitud del amoníaco y el agua significa que los dos compuestos son miscibles. De hecho, el amoníaco forma una serie de hidratos sólidos, análogos al hielo en el que los enlaces de hidrógeno definen las estructuras (Figura\(\PageIndex{3}\)). Se conocen varios hidratos de amoníaco, entre ellos: NH 3 .2H 2 O (amoniaco dihidrato, ADH), NH 3 .H 2 O (amoniaco monohidrato, AMH) y 2NH 3 .2H 2 O (hemihidrato de amoníaco, AHH).
Cabe señalar que estos hidratos no contienen iones NH 4 + u OH - discretos, lo que indica que el hidróxido amónico no existe como especie discreta a pesar del uso común del nombre. En solución acuosa, el amoníaco es una base débil (pK b = 4.75), (8.3.6).
\[ \rm NH_{3(aq)} + H_2O_{(l)} \rightleftharpoons NH_{4(aq)}^+ + OH^-_{(aq)}\]
Nota
Las soluciones de amoníaco comúnmente utilizadas en el laboratorio son una solución al 35% en agua. En clima cálido la solución desarrolla presión y la tapa debe liberarse con cuidado. La solución al 25% que se vende comercialmente (para uso doméstico) está libre de este problema.
El amoníaco es una base de Lewis y forma fácilmente complejos ácido-base de Lewis con metales de transición (8.3.7) y metales del grupo principal (Figura\(\PageIndex{4}\)).
\[ \rm [Ni(H_2O)_6]^{2+} + 6 NH_3 \rightleftharpoons [Ni(NH_3)_6]^{2+} + 6 H_2O\]
La formación de complejos estables de amoníaco es la base de un método de detección simple pero efectivo: el reactivo de Nessler, (8.3.8). Utilizando una solución de 0.09 mol/L de tetraiodomercurato de potasio (II), K 2 [HGi 4], en 2.5 mol/l de hidróxido de potasio. Una coloración amarilla indica la presencia de amoníaco: a concentraciones más altas, se puede formar un precipitado marrón. La sensibilidad como prueba puntual es de aproximadamente 0.3 μg NH 3 en 2 μL.
\[\rm NH_4^+ + 2 [HgI_4]^{2-} + 4 OH^- \rightarrow HgO\cdotHg(NH_2)I + 7 I^- + 3 H_2O\]
El amoníaco forma una solución azul con metales del Grupo 1. Como ejemplo, la disolución del sodio en amoníaco líquido da como resultado la formación de cationes Na+ solvatados y electrones, (8.3.9) donde solv = NH 3. Los electrones solvatados son estables en amoníaco líquido y forman un complejo: [e - (NH 3) 6].
\[ \rm Na_{(s)} \rightarrow Na_{(solv)} \rightleftharpoons Na^+_{(solv)} + e_{(solv)}^-\]
Es este electrón solvatado el que da las fuertes propiedades reductoras de la solución así como la señal característica en el espectro ESR asociada a un solo electrón desapareado. El color azul de la solución suele atribuirse a estos electrones solvatados; sin embargo, su absorción se encuentra en la región del infrarrojo lejano del espectro. Una segunda especie, Na - (solv), es en realidad responsable del color azul de la solución.
\[\rm 2 Na_{(solv)} \rightleftharpoons Na^+_{(solv)} + Na^-_{(solv)} \]
La reacción del amoníaco con oxígeno es altamente favorecida, (8.3.11), y el límite de inflamabilidad del amoníaco es 16 — 25 vol%. Si la reacción se lleva a cabo en presencia de un catalizador (Pt o Pd) la reacción puede limitarse a la formación de óxido nítrico (NO), (8.3.12).
\[\rm 4 NH_3 + 3 O_2 \rightarrow 2 N_2 + 6 H_2O \]
\[\rm 4 NH_3 + 5 O_2 \rightarrow 4 NO + 6 H_2O \]
Sales de amonio
El catión amonio (NH 4 +) se comporta de manera similar a los iones metálicos del Grupo 1. La solubilidad y estructura de las sales de amonio se asemeja particularmente a las del potasio y el rubidio por su tamaño relativo (Tabla\(\PageIndex{3}\)). Una diferencia es que las sales de amonio a menudo se descomponen al calentarse, (8.3.13).
\[\rm NH_4Cl_{(s)} \rightarrow NH_{3(g)} + HCl_{(g)}\]
| Cation | Radio iónico (Å) |
|---|---|
| K + | 1.33 |
| NH 4 + | 1.43 |
| Rb + | 1.47 |
La descomposición de las sales de amonio de los ácidos oxidantes a menudo puede ser violenta a altamente explosiva, y deben tratarse con cuidado. Por ejemplo, mientras que el dicromato amónico, (NH 4) 2 Cr 2 O 7, se descompone para dar un volcán (Figura\(\PageIndex{5}\)), el permanganato de amonio, NH 4 [MnO 4], es sensible a la fricción y explota a 60 °C Nitrato de amonio, NH 4 [NO 3], puede provocar incendio si se pone en contacto con un material combustible y es un ingrediente común en los explosivos ya que actúa como fuente de oxígeno debido a su balance positivo de oxígeno, es decir, el compuesto libera excedente de oxígeno a sus propias necesidades al descomponerse, (8.3.14).
\[\rm NH_4[NO_3] \rightarrow H_2O + N_2 + O\]

Bibliografía
- A. R. Barron, El detonador, 2009, 36, 60.
- A. D. Fortes, E. Suard, M. -H. Lemée-Cailleau, C. J. Pickard, y R. J. Needs, J. Am. Chem. Soc. , 2009, 131, 13508.
- M. D. Healy, J. T. Leman, y A. R. Barron, J. Am. Chem. Soc. , 1991, 113, 2776.
- A. I. Vogel y G. Svehla, Textbook of Macro and Semimicro Qualitative Inorganic Analysis, Longman, Londres (1979).
Amoníaco líquido como solvente
El amoníaco tiene un rango de líquido razonable (-77 a —33 °C), y como tal se puede licuar fácilmente con hielo seco (CO 2 sólido, T sub = -78.5 °C) y manejarse en un termo. El alto punto de ebullición del amoníaco en relación con sus congéneres más pesados es indicativo de la formación de fuertes enlaces de hidrógeno, lo que también resulta en un alto calor de vaporización (23.35 kJ/mol). Como consecuencia, el amoníaco se puede utilizar convenientemente como líquido a temperatura ambiente a pesar de su bajo punto de ebullición.
El amoníaco líquido es un buen disolvente para moléculas orgánicas (por ejemplo, ésteres, aminas, benceno y alcoholes). Es un mejor disolvente para los compuestos orgánicos que el agua, pero un peor disolvente para los compuestos inorgánicos. La solubilidad de las sales inorgánicas es altamente dependiente de la identidad del contraión (Tabla\(\PageIndex{4}\)).
| Soluble en líquido NH 3 | Generalmente insoluble en líquido NH 3 |
|---|---|
| SCN -, I -, NH 4 +, NO 3 -, NO 2 -, ClO 4 - | F -, Cl -, Br -, CO 3 2-, SO 4 2-, O 2-, OH -, S 2- |
La diferencia en la solubilidad de las sales inorgánicas en amoníaco en comparación con el agua, así como la menor temperatura del amoníaco líquido, se pueden utilizar con buena ventaja en el aislamiento de compuestos inestables. Por ejemplo, el intento de síntesis de nitrato de amonio por la reacción de nitrato de sodio y cloruro de amonio en agua da como resultado la formación de nitrógeno y agua debido a la descomposición del nitrato, (8.3.15). Por el contrario, si la reacción se lleva a cabo en amoníaco líquido, el producto secundario de cloruro de sodio es insoluble y el nitrato de amonio puede aislarse como un sólido blanco después de la filtración y evaporación por debajo de su temperatura de descomposición de 0 °C, (8.3.16).
\[ \rm NaNO_2 + NH_4Cl \xrightarrow{H_2O} NaCl + NH_4(NO_2) \rightarrow N_2 + 2 H_2O\]
\[ \rm NaNO_2 + NH_4Cl \xrightarrow{NH_3} NaCl \downarrow + NH_4(NO_2) \]
Amonación
La amonación se define como una reacción en la que se agrega amoníaco a otras moléculas o iones mediante la formación de enlaces covalentes utilizando el par de electrones no compartidos en el átomo de nitrógeno, o a través de interacciones electrostáticas ión-dipolo. En términos simples el complejo de amina resultante se forma cuando el amoníaco está actuando como una base de Lewis a un ácido de Lewis, (8.3.17) y (8.3.18), o como ligando a un catión, por ejemplo, [Pt (NH 3) 4] 2+, [Ni (NH 3) 6] 2+, [Cr (NH 3) 6] 3+, y [Co (NH 3) 6] 3+.
\[ \rm SiF_4 + 2 NH_3 \rightarrow SiF_4(NH_3)_2 \]
\[ \rm BF_3 + NH_3 \rightarrow BF_3(NH_3)\]
Ammonólisis
La amonólisis con amoníaco es una reacción análoga a la hidrólisis con agua, es decir, una reacción de disociación de la molécula de amoníaco que produce H + y una especie NH 2 -. Las reacciones de amonólisis ocurren con haluros inorgánicos, (8.3.18) y (8.3.19), y compuestos organometálicos, (8.3.20). En ambos casos, el resto NH 2 - forma un sustituyente o ligando.
\[ \rm P(O)Cl_3 + 6 NH_3 \rightarrow P(O)(NH_2)_3 + 3 NH_4Cl\]
\[ \rm BCl_3 + 6 NH_3 \rightarrow B(NH_2)_3 + 3 NH_4Cl \]
\[ \rm Al(CH_3)_3 + NH_3 \rightarrow \dfrac{1}{n}[(H_3C)_2Al(NH_2)]_n + CH_4\]
La reacción de ésteres, (8.3.21), y haluros de arilo, (8.3.22), son también ejemplos de reacciones de amonólisis.
\[ \rm RC(O)OR' + NH_3 \rightarrow RC(O)NH_2 + R'OH\]
\[ \rm C_6H_5Cl + 2 NH_3 \rightarrow C_6H_5NH_2 + NH_4Cl\]
Amidas homolépticas
Un compuesto homoléptico es un compuesto con todos los ligandos idénticos, por ejemplo, M (NH 2) n. Se logra una ruta general a compuestos de amida homolépticos mediante la reacción de una sal del metal deseado que es soluble en amoníaco líquido (Tabla\(\PageIndex{4}\)) con una amida soluble del Grupo 1. La solubilidad de las amidas del Grupo 1 se da en la Tabla\(\PageIndex{5}\). Dado que todas las amidas son insolubles (excepto las de los metales del Grupo 1) son insolubles en amoníaco líquido, la amida resultante puede aislarse fácilmente, por ejemplo, (8.3.23) y (8.3.24).
\[ \rm Mn(SCN)_2 + 2 KNH_2 \rightarrow Mn(NH_2)_2 \downarrow + 2 KSCN\]
\[ \rm Cr(NO_3)_3 + 3 KNH_2 \rightarrow Cr(NH_2)_3 \downarrow + 3 KNO_3\]
| Amida | Solubilidad en amoníaco líquido |
| LinH 2 | Discreentemente soluble |
| NanH | Discreentemente soluble |
| KNH 2 | Soluble |
| RBnH | Soluble |
| CSnH 2 | Soluble |
Reacciones redox
El amoníaco es pobre como oxidante ya que se oxida con relativa facilidad, por ejemplo, (8.3.25) y (8.3.26). Así, si es necesario realizar una reacción de oxidación el amoníaco no es un disolvente adecuado; sin embargo, es un buen disolvente para las reacciones de reducción.
\[ \rm 4 NH_3 + 5 O_2 \rightarrow 4 NO + 6 H_2O \]
\[ \rm 2 NH_3 + 3 CuO \rightarrow N_2 + 3 H_2O + 3 Cu\]
El amoníaco líquido disolverá los metales del Grupo 1 (alcalinos) y otros metales electropositivos como calcio, estroncio, bario, magnesio, aluminio, europio e iterbio. A bajas concentraciones (ca. 0.06 mol/L), se forman soluciones de azul profundo: estas contienen cationes metálicos y electrones solvatados, (8.3.27). Los electrones solvatados son estables en amoníaco líquido y forman un complejo: [e - (NH 3) 6].
\[ \rm Na_{(s)} \rightarrow Na_{(solv)} \rightleftharpoons Na^+_{(solv)} + e^-_{(solv)}\]
Los electrones solvatados proporcionan un agente reductor adecuado y potente para una gama de reacciones que normalmente no se realizan, por ejemplo, (8.3.28) y (8.3.29).
\[ \rm [Ni(CN)_4]^{2-} + 2 e^-_{(solv)} \rightarrow [Ni(CN)_4]^{4-}\]
\ [\ rm Co_2 (CO) _8 + 2 e^-_ {(solv)}\ fila derecha 2 [Co (CO) _4] ^-
El rápido aumento de la población alemana también ejerció presión sobre los recursos alimentarios de los países. Lo que agravó este tema fue que las familias aristocráticas Junkers de Prusia Oriental que poseían gran parte de la tierra en lo que se conocía como el granero de Alemania. Los junkers cultivaban centeno en sus fincas porque el suelo era demasiado ligero para el trigo, y ya que el centeno se fertilizaba con potasa (óxido de potasio, K 2 O) de la cual Alemania tenía vastos recursos. Sin embargo, en 1870 el grano de EU se estaba volviendo más barato y por lo tanto competitivo con el centeno alemán. Para proteger sus ganancias los Junkers exigieron tanto subsidios para la exportación de centeno como aranceles para la importación de trigo. El resultado de esto fue que todo el centeno alemán salía del país y no se producía suficiente trigo para satisfacer las necesidades de la población local. Se podría cultivar suficiente trigo en Alemania si se disponía de un fertilizante adecuado.
El nitrato de sodio (NaNO 3), también conocido como salitre de Chile, fue el fertilizante más común. Desafortunadamente, para 1900 los depósitos parecían estar agotados y se necesitaba una alternativa. La alternativa se encontró como componente en el alquitrán de hulla. Se supo que uno de los químicos que causaron el hedor asociado al gas de carbón y alquitrán de hulla fue el amoníaco (NH 3). El químico George Fownes (Figura\(\PageIndex{8}\)) había sugerido que el amoníaco se convirtiera en una sal y se usara como fertilizante. Desafortunadamente, la cantidad de amoníaco que se podía separar del alquitrán de hulla seguía siendo insuficiente, por lo que si el amoníaco se podía hacer a una escala lo suficientemente grande, entonces se podría realizar la fabricación a gran escala de un fertilizante.

En 1909 Fritz Haber (Figura\(\PageIndex{9}\)) presentó un método de síntesis de amoníaco a BASF. Su trabajo en colaboración con Carl Bosch (Figura\(\PageIndex{10}\)) dio como resultado el proceso conocido como el proceso Haber-Bosch en el que se mezclan nitrógeno e hidrógeno a alta temperatura (600 °C) bajo presión (200 atm) sobre un catalizador de osmio, (8.3.30).
\[ \rm N_2 + 3 H_2 \xrightarrow{\text{Os cat}} 6 NH_3 \]
Es interesante señalar que la realización del proceso Haber-Bosch requirió no solo que la industria siderúrgica construyera no solo recipientes de alta presión, sino también las formas líquidas de nitrógeno e hidrógeno. Al final resultó que el amoníaco era un componente necesario para permitir la producción de nitrógeno líquido e hidrógeno, e implicaba una falsa hipótesis de lo que causó la malaria, lo que llevó a un deseo de mantener frías las bebidas.
Mucho antes de que se entendiera la verdadera causa de la malaria, John Gorrie (Figura\(\PageIndex{11}\)), médico que trabajaba en Apalachicola en la costa del Golfo de Florida, estaba obsesionado con encontrar una cura para la malaria. El término malaria se originó en el italiano medieval: mala aria (mal aire), y se asoció con pantanos y marisma. Gorrie notó que la malaria estaba conectada al clima caluroso y húmedo por lo que comenzó a colgar un cuenco de hielo en las salas y a hacer circular el aire con un ventilador. Sin embargo, el hielo se cortó de lagos y ríos congelados en el noreste de Estados Unidos, se almacenó y luego se envió a todo el mundo, y Apalachicola era tan pequeña que rara vez se entregaba hielo. Gorrie comenzó a investigar métodos para hacer hielo. Era bien sabido que cuando un gas comprimido se expande toma calor de sus alrededores. Gorrie hizo una máquina de vapor que comprimió aire en un pistón, que cuando el pistón retraía el aire se enfriaba. En la siguiente carrera de compresión, el aire frío fue empujado hacia afuera a través de una solución de salmuera (NaCl acuoso saturado) enfriándolo. Cuando puso el agua en contacto con la salmuera fría, ésta se congeló creando el primer hielo hecho por el hombre. El 14 de julio de 1850 Gorrie produjo hielo para el Cónsul francés para enfriar el champán para la celebración del Día de la Bastilla. Justo antes de morir, Gorrie sugirió que su (para entonces) proceso patentado podría usarse para enfriar alimentos para su transporte, y fue esta aplicación la que fue ampliamente utilizada por los comerciantes británicos para llevar carne de Australia a Gran Bretaña. Sin embargo, en Alemania, la invención de Gorrie fue más útil para la cerveza.
Mientras que los británicos elaboraban cerveza tradicionalmente en la que la levadura fermenta en la superficie (fermentación superior) a una temperatura de 60 — 70 °F, en Alemania la cerveza se elaboraba utilizando fermentación de fondo. Este estilo de fermentación requiere una temperatura justo por encima de la congelación. Tradicionalmente, las bodegas frías se usaban para almacenar la cerveza fermentadora, y es de aquí el nombre lager se deriva del verbo alemán largern: almacenar. Había habido una ley en Alemania que impedía elaborar cerveza en el verano, pero con el proceso de Gorrie la posibilidad era poder elaborar cerveza todo el año. Se le pidió a Carl von Linde (Figura\(\PageIndex{12}\)) desarrollar un sistema de refrigeración. Utilizó amoníaco en lugar de aire en el sistema de Gorrie, y en 1879 estableció una empresa para comercializar sus ideas. El éxito de su refrigerador fue tal que para 1891 se estaban utilizando más de 12 mil frigoríficos, y lo que es más importante ahora existía un método conveniente para licuar gases como el hidrógeno y el nitrógeno; ambos necesarios para el proceso Haber-Bosch.
Como consecuencia del uso de amoníaco como refrigerante, fue posible preparar amoníaco a gran escala industrial. El amoníaco preparado por el proceso Haber-Bosch se puede convertir en ácido nítrico mediante el proceso Ostwald desarrollado por Wilhelm Ostwald (Figura\(\PageIndex{13}\)). El tratamiento del amoníaco con aire sobre un catalizador de platino produce inicialmente óxido nítrico, (8.3.31), y posteriormente a dióxido de nitrógeno, (8.3.32), que se disuelve en agua para dar ácido nítrico, (8.3.33).
\[ \rm 4 NH_3 + 5 O_2 \xrightarrow{\text{Pt cat}} 4 NO + 6 H_2O \]
\[ \rm 2 NO + O_2 \xrightarrow{\text{Pt cat}} 2 NO_2 \]
\[ \rm 3 NO_2 + H_2O \rightarrow 2 HNO_3 + NO\]
La adición de soda (hidróxido de sodio, NaOH) al ácido nítrico da como resultado la formación de nitrato de sodio, (8.3.34), que fue el mismo fertilizante producido a partir de los depósitos en Chile.
\[ \rm HNO_3 + NaOH \rightarrow NaNO_3 + H_2O \]

Desafortunadamente, para los procesos de Haber-Bosch y Ostwald, se sintetizó aproximadamente al mismo tiempo una forma de fertilizante aún más barata utilizando carburo de calcio para preparar cianamida cálcica (CaCN 2), (8.3.35). Como consecuencia, el proceso Haber-Bosch quedó olvidado hasta el estallido de la Primera Guerra Mundial en 1914.
\[ \rm CaC_2 + N_2 \rightarrow CaCN_2 + C\]
A las pocas semanas del brote Alemania se dio cuenta de que solo tenía suficientes explosivos para aproximadamente un año de conflicto. Esto se debió a que la principal fuente de explosivos, el nitrato de sodio era la misma fuente que daba fertilizante, es decir, Chile. Al darse cuenta de esto, la Marina Real efectivamente bloqueó las líneas de suministro. Si Alemania no encontrara otra fuente de Gran Guerra habría terminado a principios de 1916, sin embargo, se recordó que el proceso de Haber-Bosch en combinación con los procesos de Ostwald permitiría la síntesis de ácido nítrico, que al mezclarse con algodón, elaboraba nitrocelulosa (Figura\(\PageIndex{14}\)), también conocida como algodón arma, un explosivo, (8.3.36).
\[ \rm HNO_3 + C_6H_{10}O_5 \rightarrow C_6H_7(NO_2)_3O_5 + 3 H_2O \]

Como resultado de la síntesis industrial del amoníaco Alemania pudo fabricar suficientes explosivos para combatir hasta el 11 de noviembre de 1918, momento en el que casi 10 millones estaban muertos, casi 7 millones desaparecidos, y más de 21 millones resultaron heridos (Figura\(\PageIndex{15}\)).

Hidrazina
La hidrazina (N 2 H 4) es un líquido incoloro con un olor similar al amoníaco. La hidrazina tiene propiedades físicas muy cercanas al agua, con un punto de fusión de 2 °C y un punto de ebullición de 114 °C, la similitud en su química con el agua es resultado de fuertes enlaces de hidrógeno intermoleculares.
ADVERTENCIA
La hidrazina es altamente tóxica y peligrosamente inestable, y generalmente se maneja como solución acuosa por razones de seguridad. Aun así, el hidrato de hidrazina provoca quemaduras graves en la piel y los ojos. El contacto con metales de transición, sus óxidos (por ejemplo, óxido) o sales causan descomposición catalítica y posible ignición del hidrógeno desprendido. Las reacciones con oxidantes son violentas.
Síntesis
La hidrazina se fabrica a escala industrial mediante el proceso Olin Raschig utilizando la reacción de una solución de hipoclorito de sodio con amoníaco a 5 °C para formar cloramina (NH 2 Cl) e hidróxido de sodio, (8.3.37). La solución de cloraminas se hace reaccionar con amoníaco bajo presión a 130 °C, (8.3.38). El amoníaco se usa en un exceso de 33 veces.
\[ \rm NH_3 + OCl^- \rightarrow OH^- + NH_2Cl \]
\[ \rm NH_2Cl + OH^- + NH_3 \rightarrow N_2H_4 + Cl^- + H_2O \]
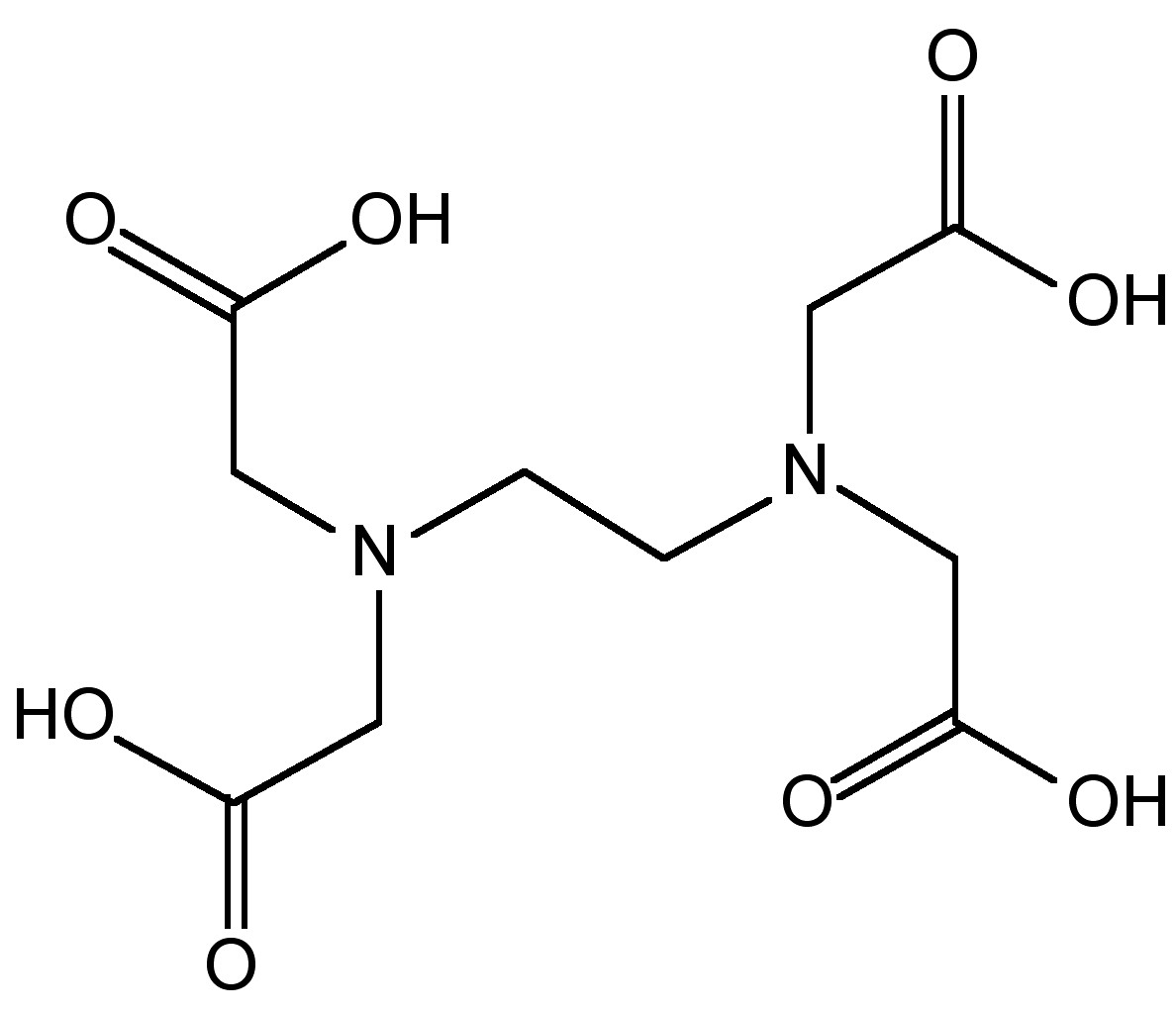
Si están presentes metales de transición entonces se produce la descomposición, (8.3.38), y por lo tanto, se agrega ácido etilendiaminotetraacético (EDTA, Figura\(\PageIndex{16}\)) para complejar los iones de metales de transición. La solución de hidrazina tal como se produce se puede concentrar por destilación para dar una solución al 65%. La hidrazina anhidra se forma por la destilación a partir de NaOH.
\[ \rm 2 NH_2Cl + N_2H_4 \rightarrow 2 NH_4^+ + 2 Cl^- + N_2\]
Las vías alternativas a la hidrazina incluyen la oxidación de la urea con hipoclorito de sodio, (8.3.40), y la reacción de amoníaco y peróxido de hidrógeno, (8.3.41).
\[ \rm (H_2N)_2\text{C=O} + NaOCl + 2 NaOH \rightarrow N_2H_4 + H_2O + NaCl + Na_2CO_3\]
\[ \rm 2 NH_3 + H_2O_2 \rightarrow N_2H_4 + 2 H_2O\]
Estructura
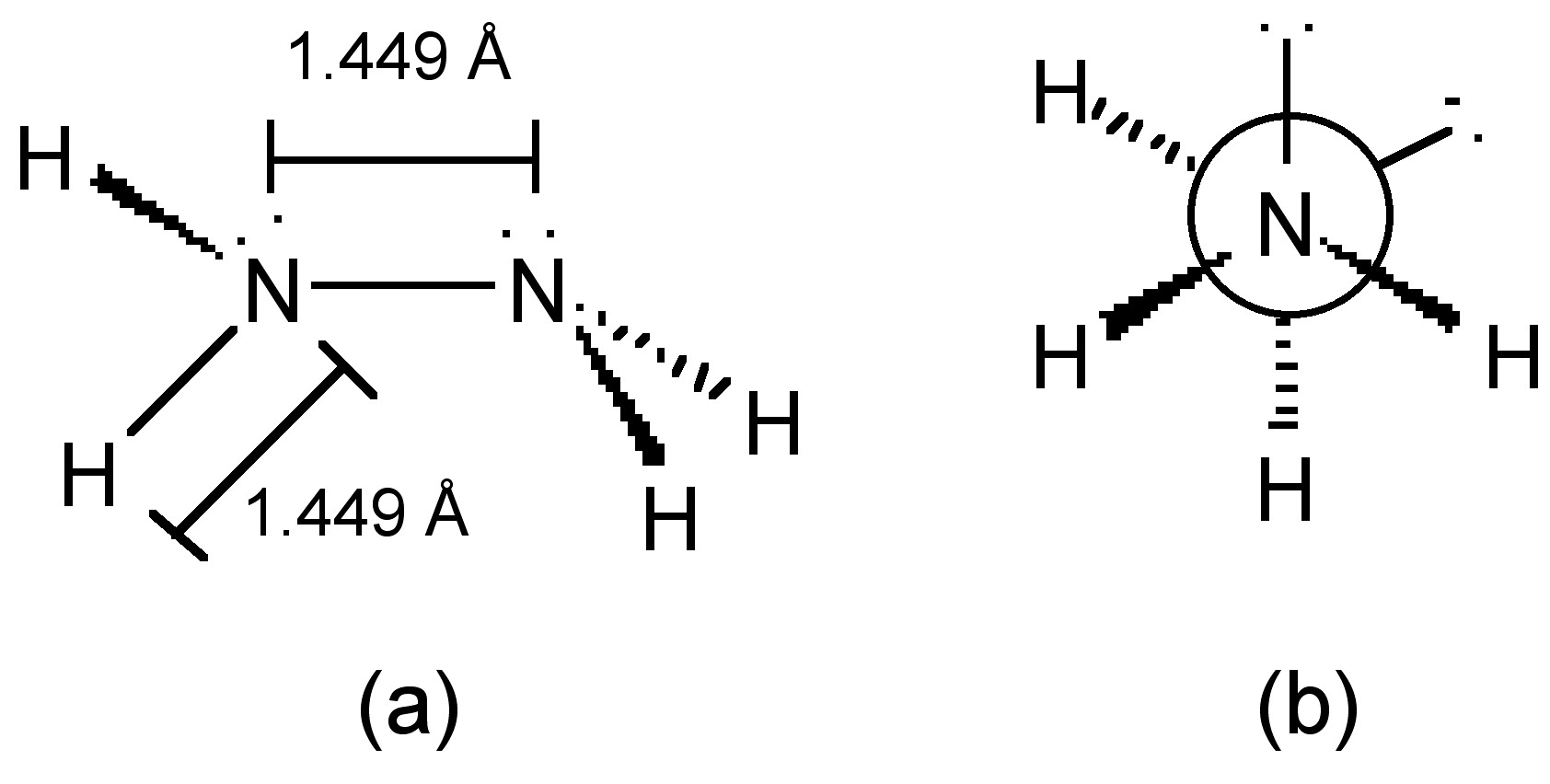
Los átomos de nitrógeno en la hidrazina adoptan hibridación sp 3 (Figura\(\PageIndex{17}\) a), y la molécula adopta una conformación gauche en los estados vapor, líquido y sólido (Figura\(\PageIndex{17}\) b).
De manera similar al amoníaco, (8.3.42), la hidrazina es una autoionizante, (8.3.43). Si bien existe una amplia gama de sales del catión N 2 H 5 +, solo las sales de sodio y potasio de N 2 H 3 - son estables.
\[ \rm 2 NH_3 \rightleftharpoons \underset{ammonium}{NH_4^+} + \underset{amide}{NH_2^-}\]
\[ \rm 2 N_2H_4 \rightleftharpoons N_2H_5^+ + N_2H_3^-\]
Química de reacción y usos
La hidrazina es polar, altamente ionizante y forma enlaces de hidrógeno estables, y su parecido con el agua se refleja en la formación de soluciones acuosas e hidratos. En estado sólido se forma el monohidrato, es decir, N 2 H 4 .H 2 O. En solución la hidrazina actúa como base para formar el ion hidrazinio, (8.3.44) donde K b = 8.5 x 10 -7. La presencia de un segundo sitio base de Lewis significa que la hidrazina puede protonarse dos veces para formar el ion hidrazonio, (8.3.45) donde K b = 8.9 x 10-16. Las sales del catión N 2 H 5 + son estables en agua; sin embargo, las sales de la dicación son menos estables.
\[ \rm 2 NH_3 \rightleftharpoons NH_4^+ + NH_2^-\]
\[ \rm N_2H_5^+ + H_2O \rightleftharpoons N_2H_5^{2+} + OH^-\]
La reacción de la hidrazina con oxígeno es altamente favorecida, (8.3.46), y el límite explosivo es 1.8 — 100 vol%.
\[ \rm N_2H_4 + O_2 \rightarrow N_2 + 2 H_2O \]
La hidrazina es útil en una serie de reacciones orgánicas para la síntesis de una amplia gama de compuestos utilizados en productos farmacéuticos, tintes textiles y en fotografía, incluyendo:
- Formación de hidrazona, (8.3.47) y (8.3.48).
- Síntesis de hidrazina alquil-sustituida mediante alquilación directa con haluros de alquilo.
- Reacción con 2-cianopiridinas para formar amida hidrazidas, las cuales pueden convertirse usando 1,2-dicetonas en triazinas.
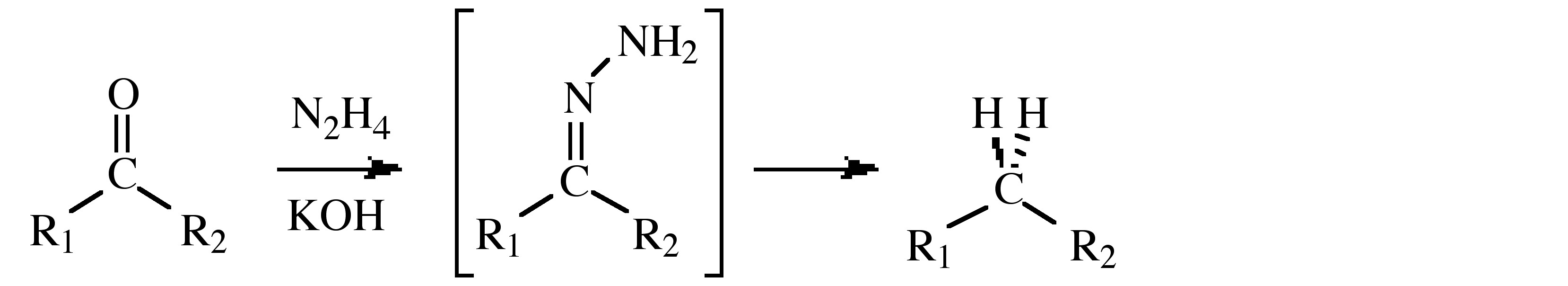
- Uso en la reducción de Wolff-Kishner que transforma el grupo carbonilo de una cetona o aldehído en un grupo metileno (o metilo) a través de un intermedio de hidrazona, (se muestra a continuación).
- Como bloque de construcción para la preparación de muchos compuestos heterocíclicos a través de la condensación con una gama de electrófilos difuncionales.
- Escisión de derivados de ftalimida N-alquilados.
- Como un reductor conveniente porque los subproductos son típicamente gas nitrógeno y agua.
\[ \rm 2 (CH_3)_2\text{C=O} + N_2H_4 \rightarrow 2 H_2O + [(CH_3)_2\text{C=N}]_2\]
\[ \rm [(CH_3)_2\text{C=N}]_2 + N_2H_4 \rightarrow 2 (CH_3)+2\text{C=N}NH_2\]
Messerschmitt Me 163 Komet
Diseñado por Alexander Lippisch (Figura\(\PageIndex{18}\)), el Messerschmitt Me 163B Komet (Figura\(\PageIndex{19}\)) fue el primer avión de combate impulsado por cohetes. Con una velocidad máxima de alrededor de 596 mph (Mach 0.83) y un techo de servicio de 40,000 pies, el rendimiento del Komet del Me 163B superó con creces al de los cazas de motor de pistón contemporáneos. Sin embargo, a pesar de su impresionante rendimiento, solo se produjo en números limitados (ca. 370 en comparación con los 1,430 construidos de su compatriota jet powered el Me 262) y no fue un avión de combate efectivo.
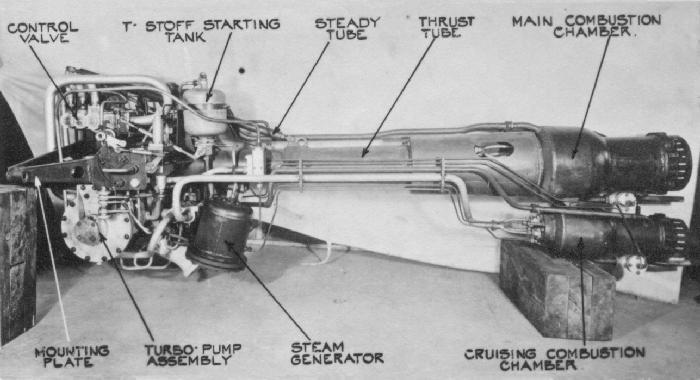
El Komet fue impulsado por el motor caliente HWK 109-509 (Figura\(\PageIndex{20}\)) que utilizó una mezcla de un combustible y un oxidante. El combustible fue una mezcla de hidrato de hidrazina (30%), metanol (57%) y agua (13%) que se designó con el nombre en clave, C-Stoff, que se quemó con el escape rico en oxígeno del peróxido de hidrógeno (T-Stoff) utilizado como oxidante. El C-Stoff se almacenó en un tanque de vidrio en el avión, mientras que el T-Stoff se almacenó en un recipiente de aluminio. Se agregó un cóctel de agente oxidante de CaMnO 4 y/o K 2 CrO 4 al T-Stoff generando vapor y altas temperaturas, esto en charrán reaccionó violentamente con el C-Stoff. El flujo de reactivos fue controlado por dos bombas, para regular la velocidad de combustión y con ello la cantidad de empuje. El violento proceso de combustión resultó en la formación de agua, dióxido de carbono y nitrógeno, y una enorme cantidad de calor enviando una corriente sobrecalentada de vapor, nitrógeno y aire que fue aspirada a través del agujero en el manto del motor, proporcionando así un empuje hacia adelante de aproximadamente 3,800 lbf. Debido a los peligros potenciales de mezclar los combustibles, se almacenaron al menos a 1/2 millas de distancia, y el avión fue lavado con agua entre los escalones de abastecimiento de combustible y después de las misiones.
Fosfina y Arsina
Debido a su uso en la deposición química de vapor de metal orgánico (MOCVD) de 13-15 (III-V) compuestos semiconductores fosfina (PH 3) y arsina (aSH 3) se preparan a escala industrial.
Síntesis
La fosfina (PH 3) se prepara mediante la reacción del fósforo elemental (P 4) con agua, (8.3.49). La fosfina ultra pura que es utilizada por la industria electrónica es preparada por la desproporción térmica del ácido fosforoso, (8.3.50).
\[ \rm 2 P_4 + 12 H_2O \rightarrow 5 PH_3 + 3 H_3PO_4\]
\[ \rm 4 H_3PO_4 \rightarrow PH_3 + 3 H_3PO_4\]
La arsina se puede preparar por la reducción del cloruro, (8.3.51) o (8.3.52). Las síntesis correspondientes también pueden ser utilizadas para la estibina y la bismutina.
\[ \rm 4 AsCl_3 + 3 LiAlH_4 \rightarrow 4 AsH_3 + 3 LiAlCl_4\]
\[ \rm 4 AsCl_3 + 3 NaBH_4 \rightarrow 4 AsH_3 + 3 NaCl + 3 BCl_3\]
La hidrólisis del fosfuro de calcio o arseniuro también puede generar los trihidruros.
Estructura
El fósforo en fosfina adopta hibridación sp 3, y así la fosfina tiene una estructura paraguas (Figura\(\PageIndex{21}\) a) debido al par solitario estereoquímicamente activo. La barrera a la inversión del paraguas (E a = 155 kJ/mol) es mucho mayor que la del amoníaco (E a = 24 kJ/mol). Poniendo esta diferencia en contexto, la tasa de inversión del amoníaco es de 10 11 mientras que la de fosfina es de 10 3. Como consecuencia es posible aislar organofosfinas quirales (PRR'R”). La arsina adopta la estructura análoga (Figura\(\PageIndex{21}\) b).
Reacciones
La fosfina es solo ligeramente soluble en agua (31.2 mg/100 mL) pero es fácilmente soluble en solventes no polares. La fosfina no actúa como un ácido ni una base en el agua; sin embargo, el intercambio de protones procede a través del ion fosfonio (PH 4 +) en soluciones ácidas y vía PH 2 - a pH alto, con constantes de equilibrio K b = 4 x 10 -28 y K a = 41.6 x 10 -29, respectivamente.
La arsina tiene solubilidad en agua similar a la de la fosfina (es decir, 70 mg/100 mL), y la Ash 3 generalmente se considera no básica, pero puede protonarse por superácidos para dar sales aislables de Ash 4 +. La arsina se oxida fácilmente en el aire, (8.3.53).
\[ \rm 2 AsH_3 + 3 O_2 \rightarrow As_2O_3 + 3 H_2O\]
La arsina reaccionará violentamente en presencia de agentes oxidantes fuertes, como permanganato de potasio, hipoclorito de sodio o ácido nítrico. La arsina se descompone en sus elementos constituyentes al calentarse a 250 - 300 °C.
Prueba de Gutzeit
La prueba de Gutzeit es la prueba característica para el arsénico e implica la reacción de arsina con Ag +. La arsina se genera por reducción de compuestos acuosos de arsénico, típicamente arsenitos, con Zn en presencia de H 2 SO 4. El Ash 3 gaseoso evolucionado se expone entonces a nitrato de plata ya sea como polvo o como solución. Con AgnO 3 sólido, Ash 3 reacciona para producir Ag 4 AsnO 3 amarillo, mientras que con una solución de AgnO 3 se forma Ag 3 As negro.
Peligros
La fosfina pura es inodora, pero la fosfina de grado técnico tiene un olor muy desagradable como ajo o pescado podrido, debido a la presencia de fosfina y difosfina sustituidas (P 2 H 4). La presencia de P 2 H 4 también provoca combustión espontánea en el aire. La fosfina es altamente tóxica; los síntomas incluyen dolor en el pecho, sensación de frialdad, vértigo, dificultad para respirar, y a mayores concentraciones daño pulmonar, convulsiones y muerte. El límite recomendado (RL) es de 0.3 ppm.
La arsina es un gas incoloro inodoro que es altamente tóxico por inhalación. Debido a la oxidación por aire, es posible oler un ligero aroma a ajo cuando la arsina está presente a aproximadamente 0.5 ppm. La arsina ataca la hemoglobina en los glóbulos rojos, provocando que sean destruidos por el cuerpo. Se producen más daños en el riñón y el hígado. La exposición a concentraciones de arsina de 250 ppm es rápidamente fatal: concentraciones de 25 a 30 ppm son fatales durante 30 min de exposición, y concentraciones de 10 ppm pueden ser fatales en tiempos de exposición más largos. Los síntomas de intoxicación aparecen después de la exposición a concentraciones de 0.5 ppm y el límite recomendado (RL) es tan bajo como 0.05 ppm.
Bibliografía
- R. Minkwitz, A. Kornath, W. Sawodny, y H. Härtner, Z. Anorg. Allg. Chem. , 1994, 620, 753.