9: Estrategias en la Síntesis de Reserpina
- Page ID
- 74203
La estructura de la reserpina se resolvió en 1953. El grupo de R.B. Woodward reportó la primera síntesis de Reserpina en 1956 (J. Am. Chem. Soc., 78, 2023, 2657 (1956); Tetraedro, 2, 1 (1958)). Su análisis académico mostró claramente aspectos del retroanálisis, que apenas estaba evolucionando en ese momento. Esta síntesis exige admiración por la forma en que utilizó el análisis conformacional y los efectos estereoelectrónicos para desarrollar con precisión los estereopuntos en este problema sumamente complejo para esa época. Reconoció que el anillo E tiene una densa matriz de 5 centros asimétricos en un anillo E de seis miembros. Su desconexión de reserpina lo llevó al intermedio clave C. Podríamos formalizar su retroanálisis como se muestra en la Figura 9.1.
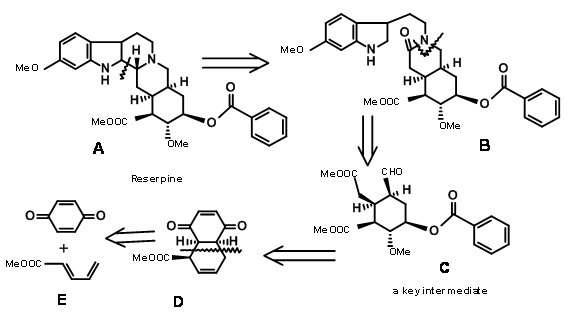
Otra pieza brillante de análisis conformacional se pudo ver en la forma en que convirtió Isoreserpina en Reserpina mediante la introducción de una cepa conformacional en una molécula por lo demás cómoda.
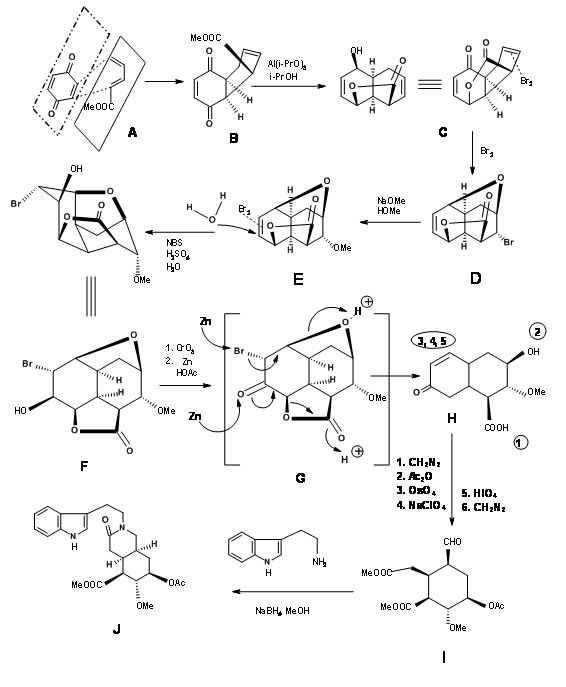
Síntesis del aldehído de Woodward (9.1C): En una ejecución de cuchilla por el grupo de Woodward (Fig 9.2), todos los carbonos requeridos para los anillos D/E y tres de los cinco centros asimétricos fueron creados por una reacción de Diels-Alder (9.2B). Tenga en cuenta que la simple asimetría en un componente, el acrilato de metilo, podría colocar con precisión tres centros asimétricos en una fila de manera correcta. Esta reacción de cicloadición desarrolló una fase cóncava y una fase convexa1 en el producto que guiaron modificaciones adicionales en este intermedio. El reactivo hidruro en la siguiente etapa liberó el hidruro de la fase convexa menos impedida, colocando el grupo -OH en la fase cóncava. Esto facilitó la formación de un anillo de lactona de cinco miembros (9.2C). El anillo alterno de lactona de seis miembros era presumiblemente más tenso. La bromación en 9.2C con bromo molecular formó el complejo de iones bromo a partir de la fase convexa, mientras que el grupo -OH pudo ingresar desde la fase cóncava para formar un éter (9.2D). El tratamiento con metóxido de sodio desplazó el bromo de la fase convexa, posiblemente por vía de eliminación — adición (9.2E). El siguiente complejo de iones bromonio procedió nuevamente de la fase convexa, con la molécula de agua entrando desde la fase cóncava para permitir una apertura trans-diaxial del complejo de bromonio (9.2F). Después de un paso de oxidación, la molécula se fijó ahora para una reacción compleja de doble eliminación, con el zinc atacando dos centros. Un ataque en el centro del bromuro abrió el anillo éter mientras que otro ataque en el grupo carbonilo abrió el anillo de lactona. Esta compleja reacción en tándem colocó los cinco centros asimétricos. Al abrir el anillo insaturado se obtuvo el intermedio clave 9.2I.
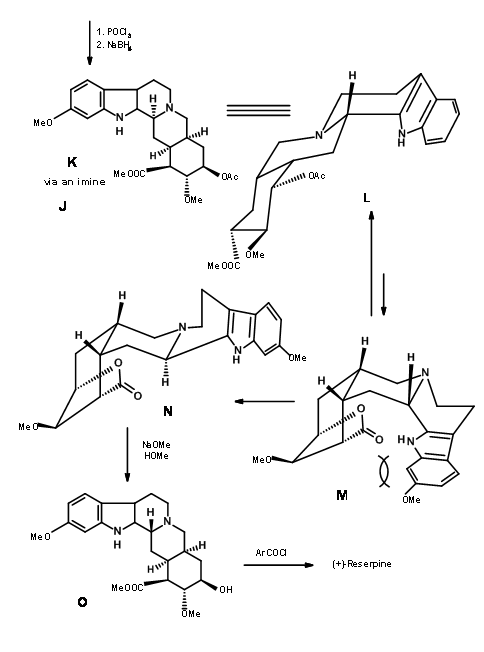
Terminación de los anillos A, B, C, D y E: La condensación de 9.2I con o-metoxitriptamina seguida de la condensación de Pictet-Spengler condujo a la formación de Isoreserpina y no Reserpina. Woodward razonó que esto se debió a que los anillos C, D y E en geometría todo-trans, tenían todos los sustituyentes en la orientación ecuatorial estable en Isoreserpina. Woodward ejecutó la isomerización en C3 de una manera ingeniosa. El álcali acuoso hidrolizó las funciones acetato-éster, el cual fue entonces lactonizado bajo condiciones DCC. Así, un anillo E inestable todo axial se bloqueó como lactona (9.2M). Esto obligó a la molécula en un estado de inestabilidad abarrotada. En la isomerización catalizada por ácido con el ácido carboxílico de alto punto de ebullición, la posición C3 se isomerizó a la configuración de reserpina. En la transesterificación, se formó el isómero correcto con un —OH libre listo para la acilación final.
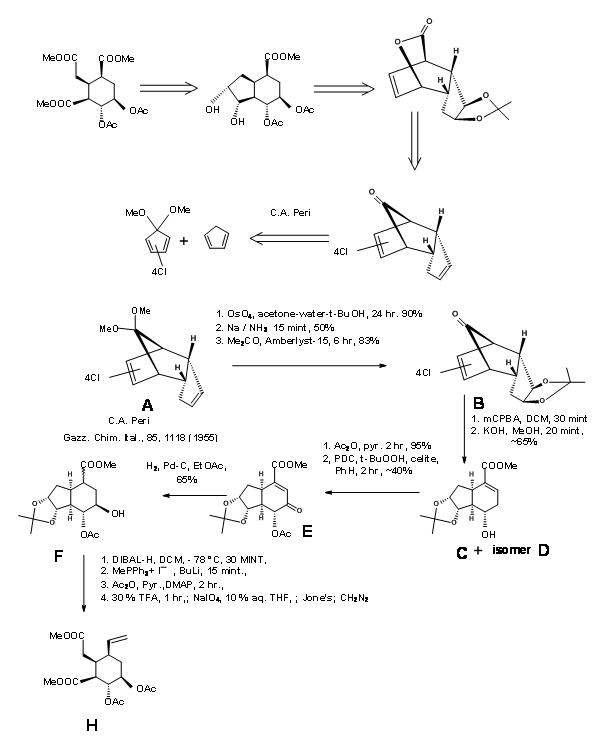
G. Metha Síntesis del anillo E: G. Metha et.al., (J. Chem. Soc., Perkin Trans. 1, 1319 (2000)) controló la estereoquímica en el anillo E a través de un sistema biciclo [2.2.1] heptano como se muestra en la Figura 9.3.
Síntesis de cigüeña: Gilbert Stork (J. Am. Chem. Soc., 127, 16255 (2005) razonó que la estereoquímica en C3 en la ruta de ciclación (AB ABDE ABCDE) de Woodward podría controlarse si se pudiera formar primero un intermedio de iminio 9.4. Esto orientaría entonces un plegamiento en forma de silla de la cadena de amarre (anillo C potencial) para un ataque axial sobre el ión iminio para dar un cierre estereoselectivo del anillo C/D para formar reserpina. Razonaron que el intermedio 9.5 serviría como intermedio clave.
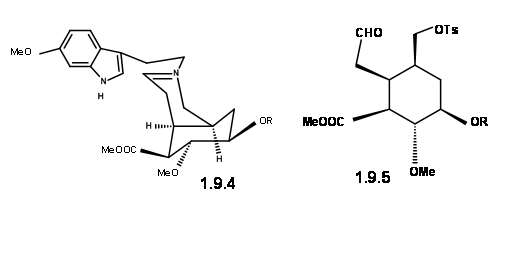
9.4 (izquierda) y 9.5 (derecha)
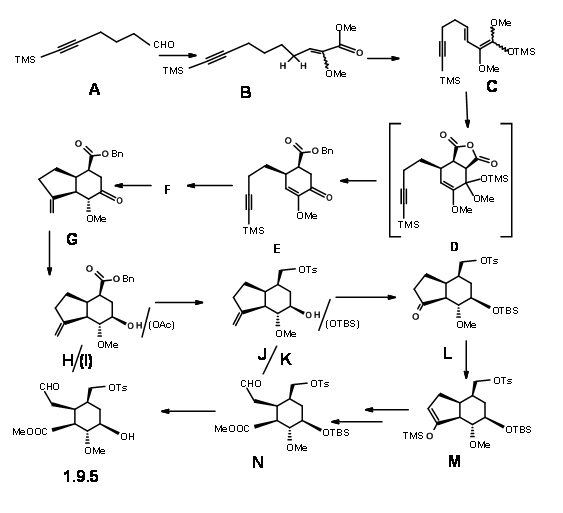
Después de algunos intentos fallidos, la escuela de Stork completó la síntesis de 9.5 utilizando una ruta que se muestra en la Figura 9.6. La condensación del hexinal 9.6A con el enolato de litio de metoxi acetato de metilo seguido de cloruro de bencenosulfonilo y posterior calentamiento con DBU dio el metoxi éster conjugado con el isómero (Z) 9.6B como producto dominante. El cloruro de trimetilsililo atrapó al dieno ceteno acetal 9.6C sin purificación del producto intermedio. La reacción de D.A con anhídrido maleico seguido de THF acuoso dio el ácido descarboxilado que se esterificó para dar 9.6E. Una ciclación de radicales libres con tributilestannano en t-butanol a reflujo y posterior tratamiento condujo a una mezcla epimérica que se equilibró con base hasta el isómero deseado 9.6G. La reducción del grupo ceto-con L-Selectride dio el alcohol axial. Este alcohol fue el invertido vía mesilación y tratamiento con acetato de cesio para dar 9.6I. La reducción con LAH, la tosilación selectiva del alcohol primario y la sililación del alcohol secundario y la ozonólisis dieron 9.6L vía 9.6K. La apertura del anillo de 5 miembros se logró atrapando el enolato cinético como derivado de TMS 9.6M y la ozonólisis y esterificación final dieron 9.6N. Este intermedio clave también se sintetizó a través de una ruta alternativa más eficiente.
Desafortunadamente, la condensación de 9.5 con el precursor de indol dio isoreserpina como el producto principal (Fig 9.7). Los investigadores razonaron que este inesperado resultado probablemente se debió a la formación del
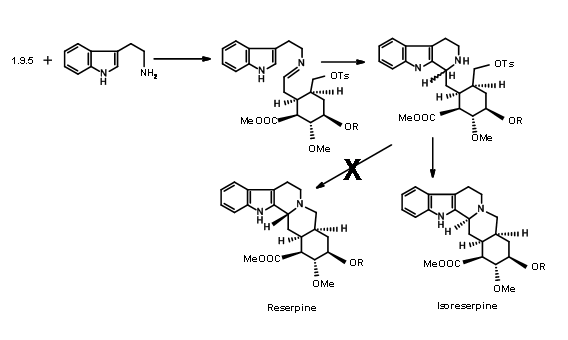
Anillo C previo a la ciclación. Para conducir la reacción hacia una formación de anillo DE, decidieron atrapar la imina intermedia como la cianoamina 9.8A. Esto se logró añadiendo un exceso de cianuro de potasio a la mezcla de reacción
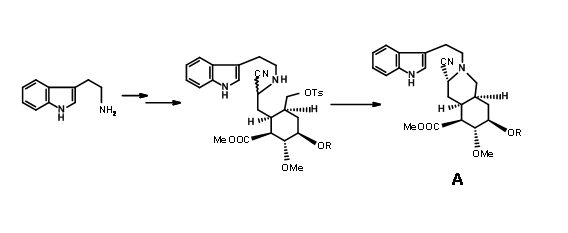
El producto esperado 9.8A con un grupo ciano axial fue el único producto. El reflujo del aminonitrilo en acetonitrilo como disolvente de nuevo dio (±) precursor de metil isoreserpina como el producto principal (Fig 9.9). Dado que el plegamiento en forma de silla esperado y un ataque axial del resto indol en el resto de ión iminio parecían estar en motivos srereoelectrónicos sólidos, los autores razonaron que el anión cianuro en la fase α de la molécula formó un par de iones estrechos con el ion imonio
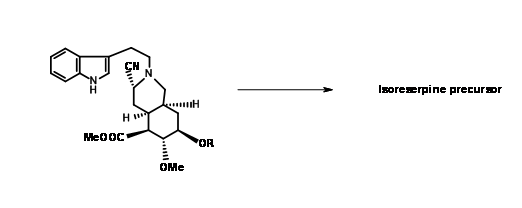
bajo estas condiciones.Este anión impidió el ataque axial sobre un plegado de la cadena en forma de silla, forzando una conformación similar a una embarcación antes de la ciclación seguido de un ataque axial para dar isoreserpina como un producto importante.
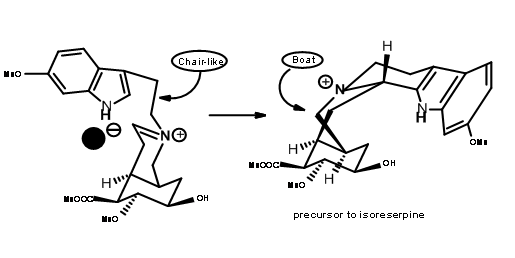
En tal caso, permitir que el contraión cianuro escape de la influencia del ión iminio mediante el uso de un disolvente polar debería limpiar el sitio de reacción para un ataque axial-silla. Cuando se trató la nitrilamina 9.8A 10%
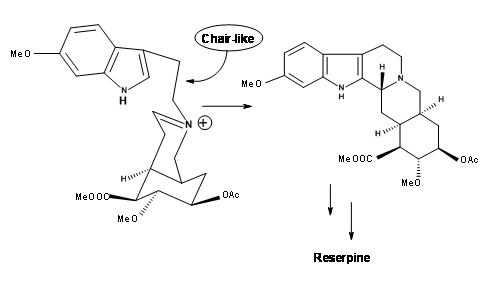
de HCl 1 N en disolvente THF, el precursor para (±) metil reserpina fue el producto con 90% de rendimiento. Esto finalmente se convirtió en reserpina por procedimientos conocidos. Con base en estos resultados, los autores han reportado otra ruta quiral muy corta. Así, la persistencia y el razonamiento mecanicista crítico en cada punto del fracaso llevaron a una síntesis directa exitosa del isómero correcto.
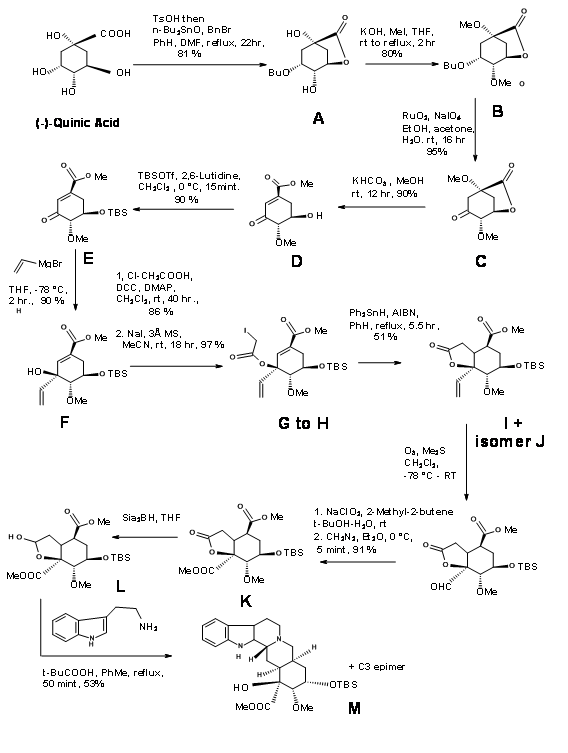
S. Hanessean et.al., (J.Org. Chem.,62, 465 (1997) aplicó el enfoque Quirón al intermedio clave 9.10B adecuado para la formación de anillos CD en un enfoque E ABCDE ABE discutido hasta ahora.
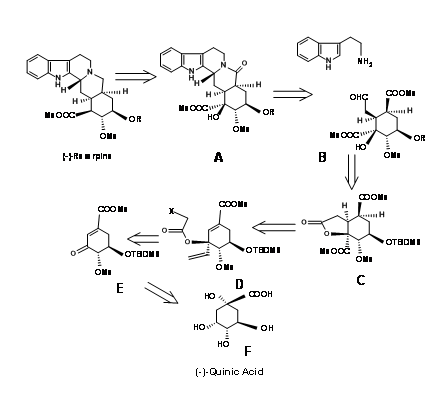
El ácido quínico 9.10F tuvo la mayoría de los centros quirales en la orientación adecuada. Sin embargo, necesitó modificación en los centros potenciales C20, C15 y C16. El retroanálisis de Hanessian se muestra en la Figura 9.10. La síntesis real se muestra en la Figura 9.11. La lactonización, seguida de la protección selectiva de bencilo y la metilación de los grupos OH restantes dieron el compuesto B. La oxidación fue seguida de metanolisis del anillo de lactona condujo a la eliminación para dar la cetona conjugada D. Protección del C18 — OH como TBDMS seguido de vinilación usando reactivo de Grignard colocado la función potencial del ácido carboxílico. El t-alcohol así generado proporcionó un anclaje a la introducción estereoespecífica de la cadena de dos carbonos necesaria. Una ciclación reductora de radicales libres colocó la cadena carbonada en la estereoquímica correcta. El isómero correcto en C20 (I) se movió luego a la siguiente etapa. Este intermedio clave L condujo a los precursores de reserpina e isoreserpina 9.11M y 9.11M 'en la proporción 1.4:1 respectivamente. Estos pasos se muestran en la Figura 9.11.