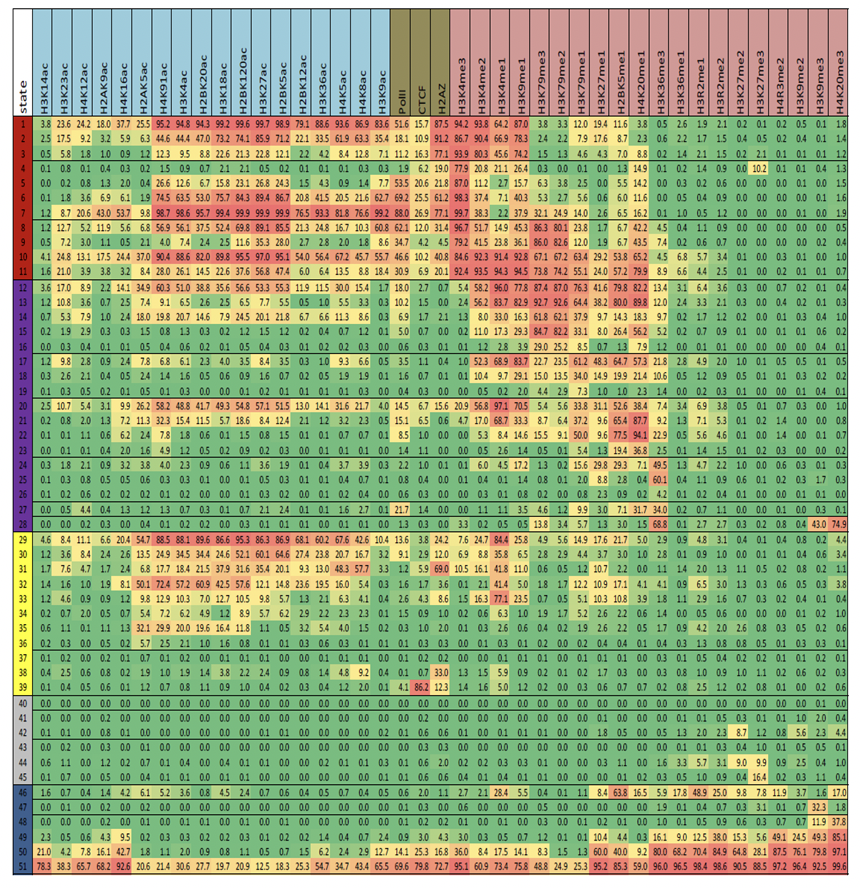
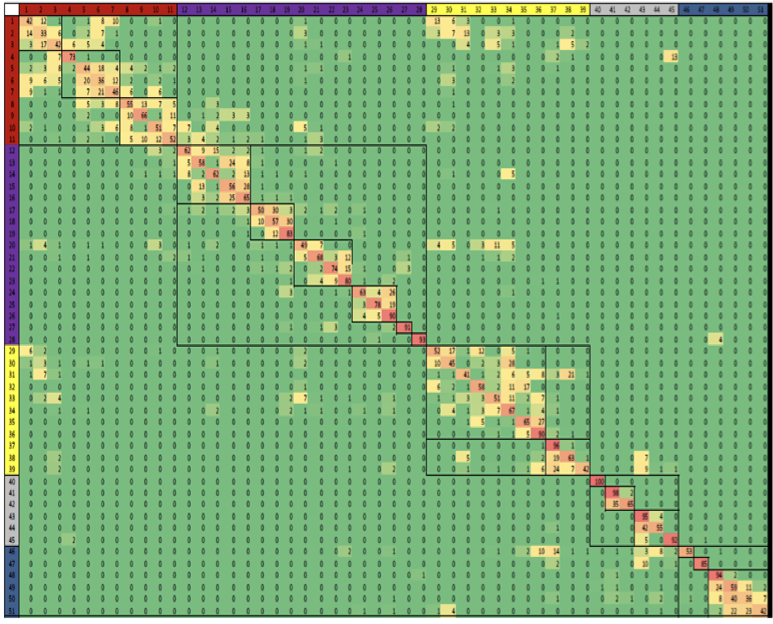
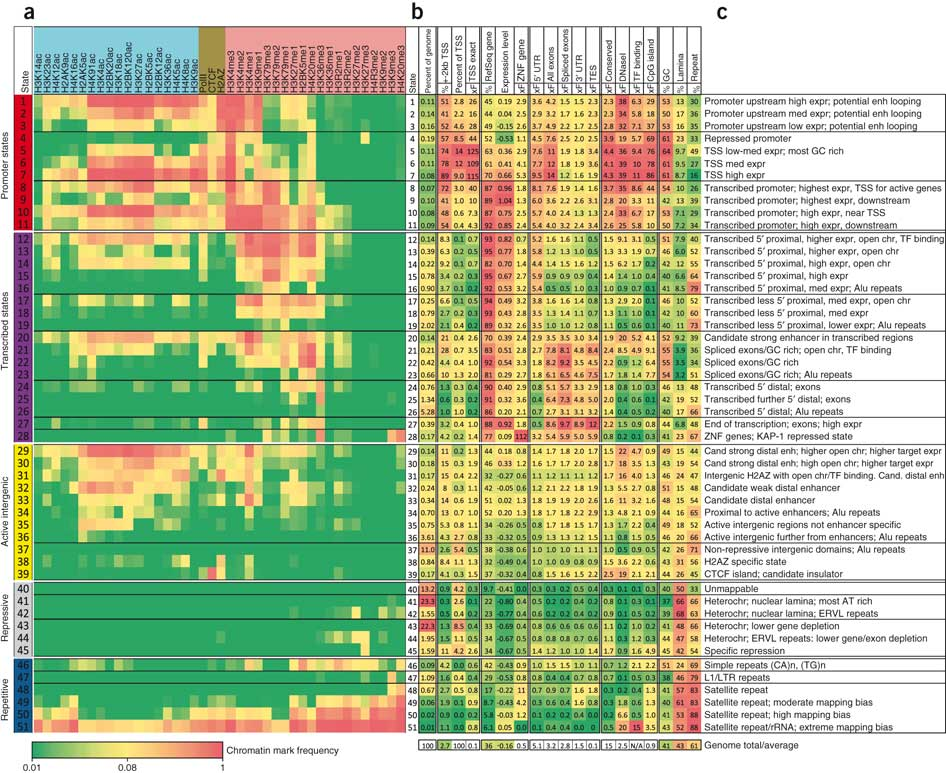
19.8: ¿Qué hemos aprendido? , Bibliografía
- Page ID
- 54038
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)En esta conferencia aprendimos cómo las marcas de cromatina pueden ser utilizadas para inferir estados biológicamente relevantes. El análisis en [7] presenta un método sofisticado para aplicar técnicas previamente aprendidas como los HMM a un problema complejo. La conferencia también presentó la poderosa transformación Burrows-Wheeler que ha permitido el mapeo de lectura eciente.
Bibliografía
[1] Langmead B, Trapnell C, Pop M y Salzberg S. Ultrafast, alineamiento memory-ecient de secuencias cortas de ADN con el genoma humano. Biología del Genoma, 10 (3), 2009.
[2] Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, et al. Análisis integrador de 111 epigenomas humanos de referencia. Naturaleza, 518 (7539) :317—330, 2015.
[3] El Consorcio del Proyecto ENCODE. Una enciclopedia integrada de elementos de ADN en el genoma humano. Naturaleza, 489 (7414) :57—74, 2012.
[4] Escuché E y Martienssen RA. Herencia epigenética transgeneracional: Mitos y mecanismos. Cell, 157 (1) :95—109, 2014.
[5] Mardis ER. Chip-seq: bienvenido a la nueva frontera. Nature Methods, 4 (8) :614—614, 2007.
[6] Herz H-M, Hu D y Shilatifard A. Mal funcionamiento del potenciador en cáncer. Molecular Cell, 53 (6) :859—866, 2014.
[7] Ernst J y Kellis M. Descubrimiento y caracterización de estados de cromatina para la anotación sistemática del genoma humano. Nature Biotechnology, 28:817 —825, 2010.
[8] Ernst J, Kheradpour P, Mikkelsen TS, et al. Mapeo y análisis de la dinámica del estado de la cromatina en nueve tipos celulares humanos. Naturaleza, 473 (7345) :43—49, 2011.
[9] Mousavi K, Zare H, dell'Orso S, Grontved L, et al. ERnas promueven la transcripción estableciendo accesibilidad a la cromatina en loci genómicos definidos. Molecular Cell, 51 (5) :606—17, 2013.
[10] Qunhua Li, James B. Brown, Haiyan Huang y Peter J. Bickel. Medición de la reproducibilidad de experimentos de alto rendimiento. Los anales de la estadística aplicada, 5 (3) :1752—1779, 2011.
[11] Li Y y Tollefsbol TO. Detección de metilación del ADN: Análisis de secuenciación genómica con bisulfito. Métodos Biología Molecular, 791:11 —21, 2011.
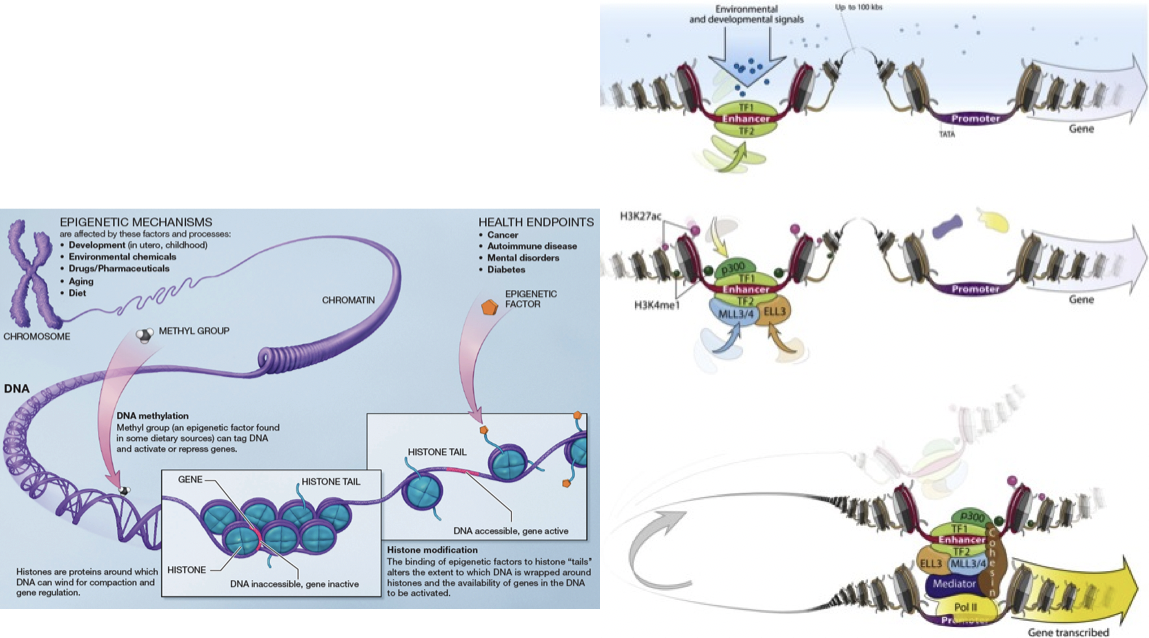
Cortesía de Elsevier, Inc. Usado con permiso. Fuente: Herz, Hans-Martin, Deqing Hu, et al. “Mal Mal Mejorador en Cáncer”. Molecular Cell 53, núm. 6 (2014): 859-66 (derecha).
Figura 19.1: A. Existe una amplia diversidad de modificaciones en el epigenoma. Algunas regiones del ADN se enrollan de forma compacta alrededor de las histonas, haciendo que el ADN sea inaccesible y los genes inactivos. Otras regiones tienen ADN más accesible y, por lo tanto, genes activos. Los factores epigenéticos pueden unirse a las colas de estas histonas para modificar estas propiedades. B. Las modificaciones de histonas proporcionan información sobre qué tipos de proteínas están unidas al ADN y cuál es la función de la región. En este ejemplo, las modificaciones de histonas permiten que una región potenciadora (potencialmente a más de 100 bases de kilo de distancia) interactúe con la región promotora. [6]
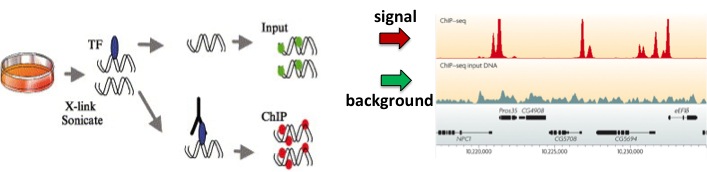
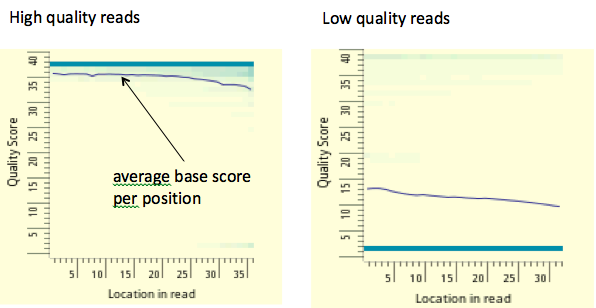
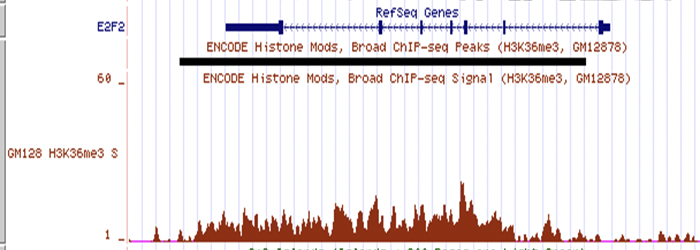
Figura 19.6: Una pista de señal de muestra. Aquí, la señal roja se deriva del número de lecturas que mapearon al genoma en cada posición para un experimento ChIP-seq con la diana H3K36me3. La señal da un nivel de enriquecimiento de la marca
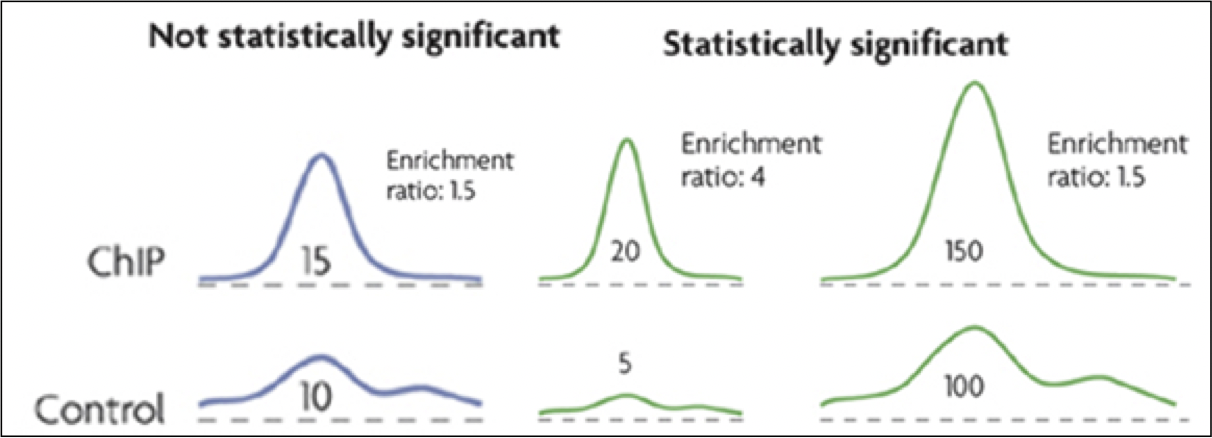
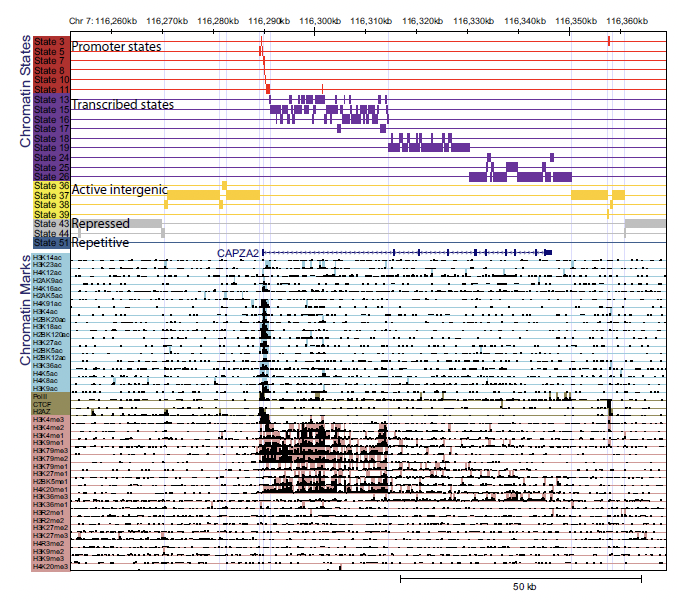
Figura 19.8: Ejemplo de los datos y la anotación del modelo HMM. La sección inferior muestra el número bruto de lecturas mapeadas al genoma. La sección superior muestra la anotación del modelo HMM.