
8.11: Secuenciación de Próxima Generación
- Page ID
- 56285

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Expansión de la tecnología de secuenciación
Crédito: Jeremy Seto (CC-BY-NC-SA 3.0)
La secuenciación tradicional de genomas fue un proceso largo y tedioso que clonó fragmentos de ADN genómico en plásmidos para generar una biblioteca de ADN genómico (ADNg). Estos plásmidos se secuenciaron individualmente utilizando la metodología de secuenciación de Sanger y se realizó computacional para identificar piezas superpuestas, como un rompecabezas. Este montaje resultaría en un andamio de tiro.
A medida que la tecnología mejoraba, el costo de secuenciar genomas se hizo menos costoso. Esta tecnología superó a la Ley de Moore, una proyección de semiconductores sobre la velocidad de las computadoras a medida que avanzaba el tiempo. Una dramática disminución del precio en el costo de la secuenciación del genoma ocurrió alrededor de 2008 debido a los avances técnicos.

A medida que el costo de la secuenciación del genoma disminuyó, se observó un aumento dramático en la deposición del genoma en Genbank. Estos depósitos reflejaban pequeños genomas de bacterias y arqueas.

Crédito: Estevezj (CC-BY-SA 3.0)
La disminución en el costo de secuenciación por nucleótido provino de la paralelización de la secuenciación. Mientras que Sanger Sequencing es capaz de secuenciar un tramo a la vez, un ensamblaje paralelo de reacciones de secuenciación ha llevado a la secuenciación de alto rendimiento a menudo llamada Next Generation Sequencing (NGS).
La próxima generación de secuenciación: tecnologías de alto rendimiento
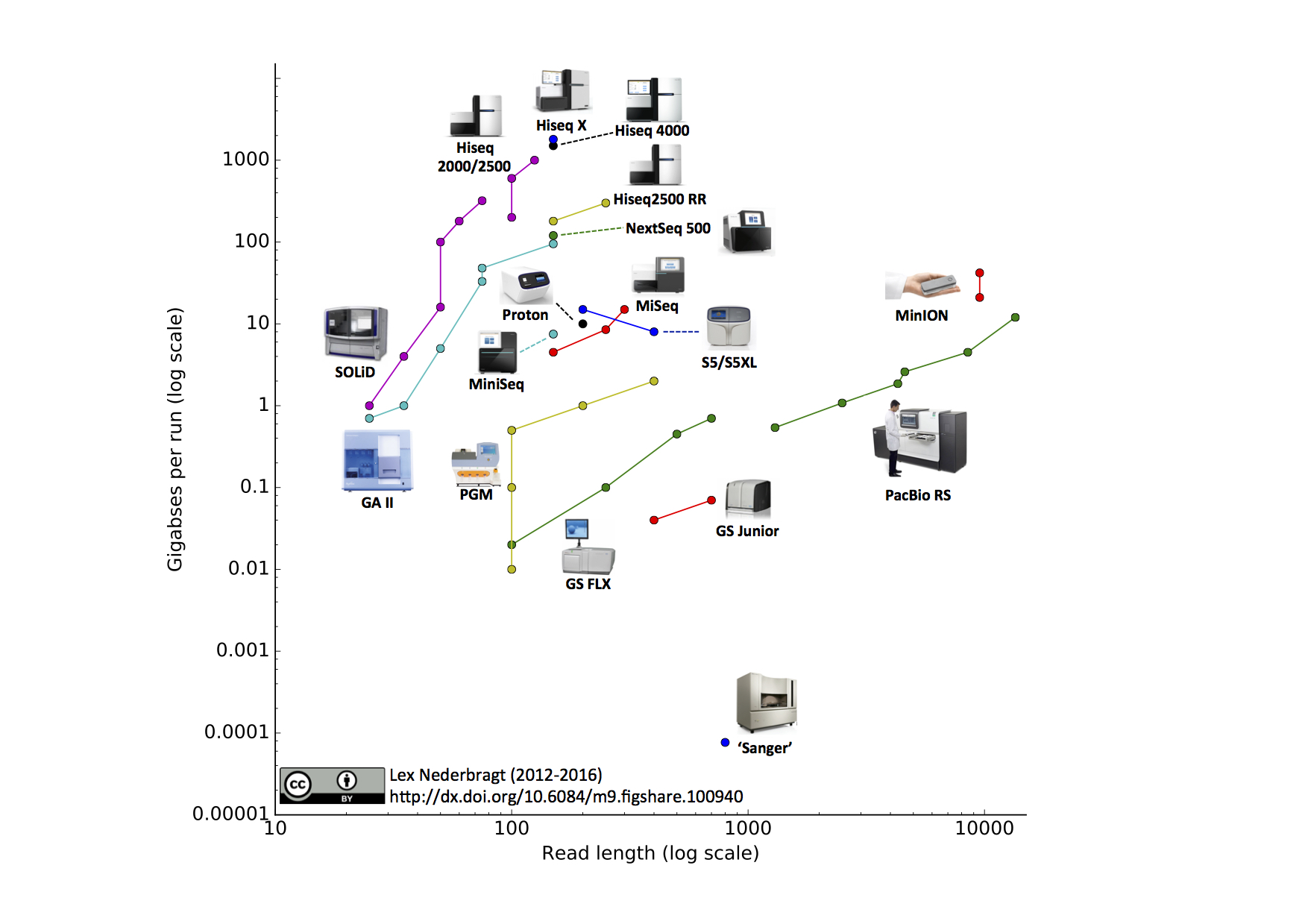
Crédito: Lex Nederbragt (CC-BY 3.0)
Secuenciación de Alto Rendimiento Aplicada a la Secuenciación Genómica (TEDED CC BY-NC-ND
Secuenciación de lectura corta por síntesis
Illumina
La secuenciación de lectura corta Illumina utiliza tecnología de células de flujo donde los oligonucleótidos complementarios a los cebadores adaptadores se siembran físicamente.

Superficie de la celda de flujo con los oligonucleótidos adaptadores. Crédito: DmLapato (CC-BY-SA 4.0)
Las secuencias de ADN fragmentadas se adaptan con cebadores mediante ligación y se hibridan con la celda de flujo. Para aumentar la señal de la secuenciación, las secuencias cortas de ADN se amplifican a través de un proceso llamado amplificación puente o generación de conglomerados.

Generación de clústeres mediante amplificación de puente. Se utiliza un er entumecido bajo de ciclos de PCR. La generación de clústeres ayuda en la posterior determinación de señal/ruido.
Crédito: DmLapato (CC-BY-SA 4.0)
La celda de flujo se somete a sucesivas rondas de inundación con un nucleótido fluorescente, se le permite incorporarse con una ADN polimerasa y se elimina por lavado. Después de cada ciclo de inundación/lavado, se miden las señales fluorescentes para indicar la incorporación. Las localizaciones específicas de fluorescencia son rastreadas y consolidadas para indicar la secuencia en cada punto registrado.

Cada ciclo de flujo introduce un nucleótido fluorescente para su incorporación. Crédito: Abizar Lakdawalla (CC-BY 3.0)
Torrente Ion
El ADN fragmentado se liga a secuencias adaptadoras y se adhiere a microperlas. Las perlas se incrustan en micropocillos en un semiconductor. Ion Torrent realiza las reacciones de secuenciación en una solución no tamponada ya que el semiconductor actúa como un medidor de pH para identificar la incorporación de nucleótidos. Los nucleótidos estándar se inundan en el chip y se incorporan. Debido a que la incorporación de nucleótidos crea un protón (H +), se detecta un microambiente de pH bajo en la solución no tamponada.
Secuenciación de una sola molécula
Pac Bio
Pac Bio utiliza nano pozos con ADN polimerasa unida por covalentía para secuenciar moléculas individuales de ADN. Los nucleótidos fluorescentes se incorporan durante las reacciones de síntesis y se puede medir la incorporación en tiempo real. La secuenciación Pac Bio tiene la ventaja de secuenciar fragmentos de 10-20kb, en marcado contraste con los métodos de lectura corta.
Nanoporo Oxford

Crédito: George Church (CC-BY 3.0)
Oxford Nanopore utiliza la proteína alfa-hemolisina integrada en un chip semiconductor. El tamaño de poro de la proteína es el tamaño correcto para que una sola molécula de ADN se ajuste a través. Una molécula de ADN polimerasa se une a la abertura del poro por donde se alimenta el ADN replicado. A medida que el ADN atraviesa el poro, los cambios de voltaje se miden y mapean a las cualidades de bases específicas.
Enlace: Youtu.be/BNZ880V52RQ
Secuencia de salida
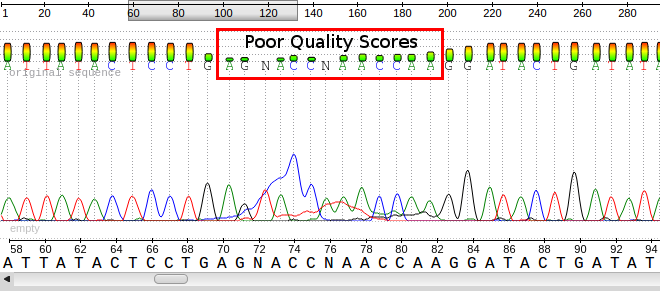
Un archivo ab1 de muestra que muestra las llamadas base, los cromatogramas y las puntuaciones de calidad para cada base. Observe la mala calidad en la caja roja y los picos/bases correspondientes.
El archivo de salida de los métodos de secuenciación de próxima generación utiliza el formato fastq. Al igual que un archivo fasta, hay un encabezado que describe la secuencia. La primera línea es la cabecera o línea de título que comienza con '@' (recuerde que fasta comienza con '>'). La segunda línea es la secuencia cruda real (una vez más similar a fasta). La tercera línea no tiene sentido mientras que la cuarta línea está llena de símbolos siempre que la línea de secuencia. Esta última línea es la puntuación de calidad de la llamada base. Al igual que con la secuenciación de Sanger, puede haber ambigüedad con la llamada base de la secuencia y la certeza se mantiene en la puntuación de calidad.
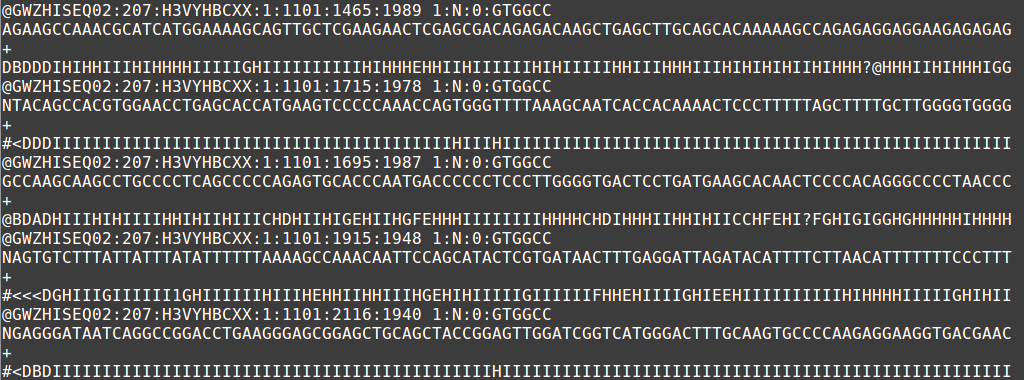
Ejemplo de archivo fastq que muestra 5 secuencias de lectura corta.
Se desarrollaron puntuaciones Phred para evaluar la calidad de las llamadas base derivadas de la secuenciación fluorescente de Sanger durante el Proyecto Genoma Humano. El programa phred escanea los picos del cromatograma y las puntuaciones en función de la certeza o precisión de la llamada. Las puntuaciones se basan logarítmicamente y las puntuaciones mayores de 20 representan una precisión mayor del 99% de la llamada base.
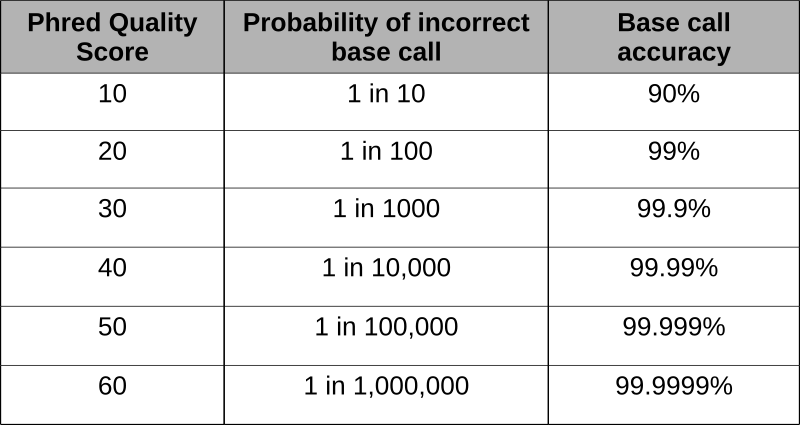
Usando las puntuaciones phred incrustadas en la última línea de archivos fastq, se pueden eliminar lecturas de mala calidad. El uso de un programa como FastQC permite la evaluación de las lecturas y produce la representación gráfica de la calidad.
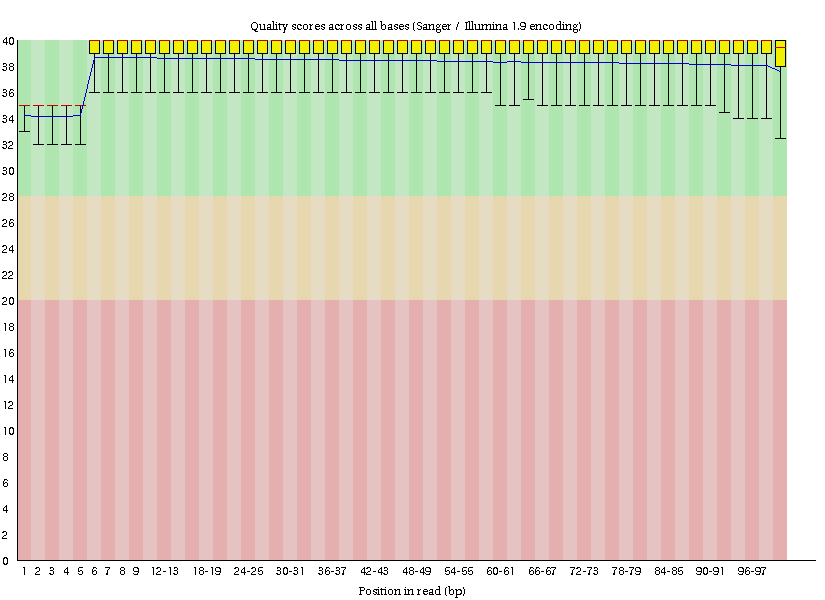
Salida de calidad FastQC que ilustra la puntuación Phred para cada llamada base.
Esta secuencia de lectura corta de aproximadamente 100 nucleótidos tiene todas las bases hechas con una precisión mayor de 30, o > 99.9%.
Ensamblaje y Alineación
Las secuencias de lecturas cortas deben ensamblarse en una secuencia utilizable. Para ello, un genoma de referencia puede ayudar en el ensamblaje después de recortar las secuencias adaptadoras usando métodos automatizados. En el caso de que no exista un genoma de referencia, se podrá utilizar una especie relacionada o se debe llevar a cabo un proceso de ensamblaje de novo más intensivo computacionalmente. Con el ensamblaje de novo, puede ser útil tener algunas lecturas largas realizadas con PacBio para crear andamios para generar el ensamblaje en secuencias contiguas, o cóntigos.

RNA-seq

Crédito: Malachi Griffith, Jason R. Walker, Nicholas C. Spies, Benjamin J. Ainscough, Obi L. Griffith (CC-BY 4.0) https://doi.org/10.1371/journal.pcbi.1004393

Crédito: Rgocs (CC-BY)
La RT-PCR y la RT-qPCR se pueden usar para medir la abundancia de transcritos específicos de una manera de rendimiento bastante bajo. Aprovechando el concepto de transcripción inversa y acoplándolo a tecnologías de secuenciación de alto rendimiento, los transcritos pueden secuenciarse y mapearse a un genoma para representar la cantidad de transcritos representados por el número de lecturas.
Dada la cobertura de lectura suficiente, también se pueden identificar nuevas isoformas de empalme, ya que se identifican diferentes uniones exón-exón.
El flujo de trabajo general del análisis de RNA-seq sigue:

Crédito: Toxina Saludable (CC-BY-SA 4.0)