
2.3: Estructura y Función- Proteínas I
- Page ID
- 52913

Fuente: BiochemFFA_2_2.pdf. Todo el libro de texto está disponible de forma gratuita de los autores en http://biochem.science.oregonstate.edu/content/biochemistry-free-and-easy
Las proteínas son los caballos de batalla de la célula. Prácticamente todo lo que sucede dentro de las células sucede como resultado de las acciones de las proteínas. Entre otras cosas, las enzimas proteicas catalizan la gran mayoría de las reacciones celulares, median en la señalización, dan estructura tanto a las células como a los organismos multicelulares, y ejercen control sobre la expresión de los genes. La vida, como la conocemos, no existiría si no hubiera proteínas. La versatilidad de las proteínas surge por sus variadas estructuras.
Las proteínas se elaboran uniendo aminoácidos, teniendo cada proteína una secuencia de aminoácidos característica y única. Para tener una idea de la diversidad de proteínas que se pueden hacer usando 20 aminoácidos diferentes, considere que el número de diferentes combinaciones posibles con 20 aminoácidos es de 20 n, donde n=el número de aminoácidos en la cadena. Se hace evidente que incluso un dipéptido hecho de apenas dos aminoácidos unidos entre sí nos da 20 2 = 400 combinaciones diferentes. Si hacemos el cálculo para un péptido corto de 10 aminoácidos, llegamos a una enorme combinación de 10,240,000,000,000. La mayoría de las proteínas son mucho más grandes que esta, lo que hace que el número posible de proteínas con secuencias de aminoácidos únicas sea inimaginablemente enorme.
Niveles de Estructura
La importancia de la secuencia única, u orden, de los aminoácidos, conocida como estructura primaria de la proteína, es que dicta la conformación 3-D que tendrá la proteína plegada. Esta conformación, a su vez, determinará la función de la proteína. Examinaremos la estructura de la proteína a cuatro niveles distintos (Figura 2.17) - 1) cómo la secuencia de los aminoácidos en una proteína (estructura primaria) da identidad y características a una proteína (Figura 2.18); 2) cómo las interacciones locales entre una parte de la cadena principal del polipéptido y otra afectan la forma de la proteína ( estructura secundaria); 3) cómo la cadena polipeptídica de una proteína puede plegarse para permitir que los aminoácidos interactúen entre sí que no están cerca en la estructura primaria (estructura terciaria); y 4) cómo diferentes cadenas polipeptídicas interactúan entre sí dentro de una proteína de múltiples subunidades (estructura cuaternaria).
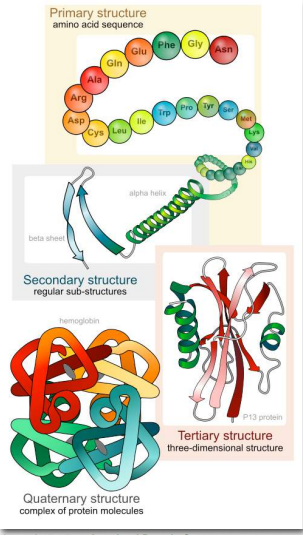
En este punto, debemos proporcionar un par de definiciones. Utilizamos el término polipéptido para referirnos a un solo polímero de aminoácidos. Puede o no haberse plegado en su forma final y funcional. El término proteína a veces se usa indistintamente con polipéptido, como en “síntesis de proteínas”. Generalmente se usa, sin embargo, para referirse a una molécula plegada funcional que puede tener una o más subunidades (compuestas por polipéptidos individuales). Así, cuando usamos el término proteína, usualmente nos referimos a un polipéptido o péptidos funcionales, plegados. La estructura es esencial para la función. Si alteras la estructura, alteras la función -por lo general, pero no siempre, esto significa que pierdes toda la función. Para muchas proteínas, no es difícil alterar la estructura.
Las proteínas son flexibles, no rígidamente fijadas en su estructura. Como veremos, es la flexibilidad de las proteínas lo que les permite ser catalizadores asombrosos y les permite adaptarse, responder y transmitir señales tras la unión de otras moléculas o proteínas. Sin embargo, las proteínas no son infinitamente flexibles. Hay restricciones en las conformaciones que las proteínas pueden adoptar y estas restricciones gobiernan las conformaciones que muestran las proteínas.
Cambios sutiles
Incluso cambios muy pequeños y sutiles en la estructura de las proteínas pueden dar lugar a grandes cambios en el comportamiento de las proteínas. La hemoglobina, por ejemplo, sufre un cambio estructural increíblemente pequeño al unirse una molécula de oxígeno, y ese simple cambio hace que el resto de la proteína gane una afinidad considerablemente mayor por el oxígeno que la proteína no tenía antes del cambio estructural.
Secuencia, estructura y función
Como se discutió anteriormente, el número de diferentes secuencias de aminoácidos posibles, incluso para péptidos cortos, es muy grande. No hay dos proteínas con diferentes secuencias de aminoácidos (estructura primaria) que tengan una estructura global idéntica. La secuencia de aminoácidos única de una proteína se refleja en su estructura plegada única. Esta estructura, a su vez, determina la función de la proteína. Es por ello que las mutaciones que alteran la secuencia de aminoácidos pueden afectar la función de una proteína.
Síntesis de Proteínas
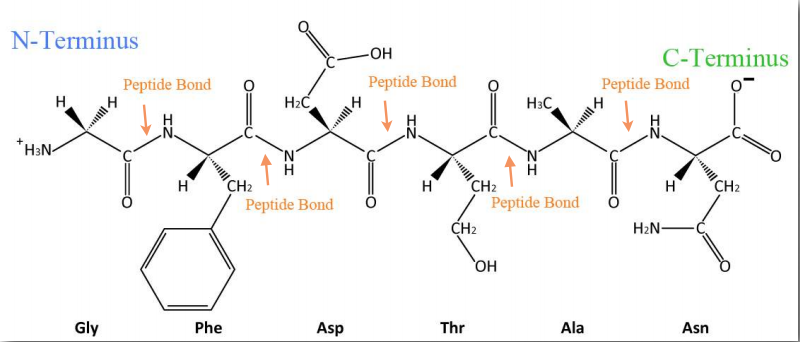
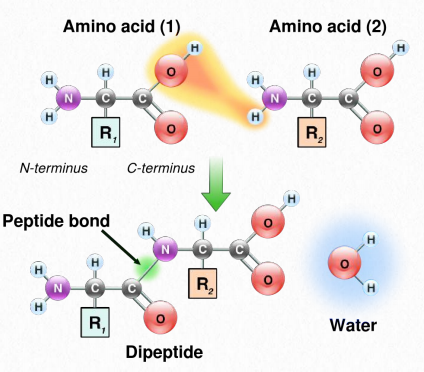
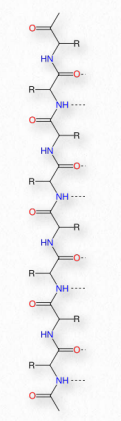
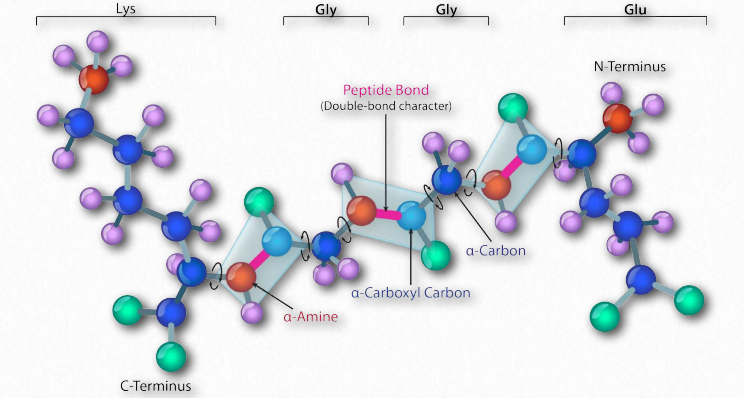
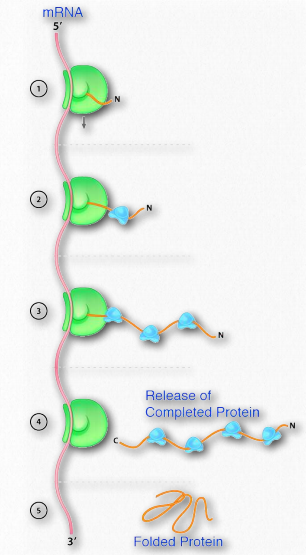
La síntesis de proteínas ocurre en los ribosomas y procede uniendo el extremo carboxilo del primer aminoácido al extremo amino del siguiente (Figura 2.19). El extremo de la proteína que tiene el grupo α-amino libre se conoce como el extremo amino o N-terminal. El otro extremo se denomina término carboxilo o extremo C, ya que contiene el único grupo α-carboxilo libre. Todos los demás grupos α-amino y grupos α-carboxilo están ligados en la formación del péptido Figura 2.19 Vinculación de aminoácidos a través de enlaces de formación de enlaces peptídicos que unen aminoácidos adyacentes entre sí. Las proteínas se sintetizan comenzando con el extremo amino terminal y terminando en el extremo carboxilo.
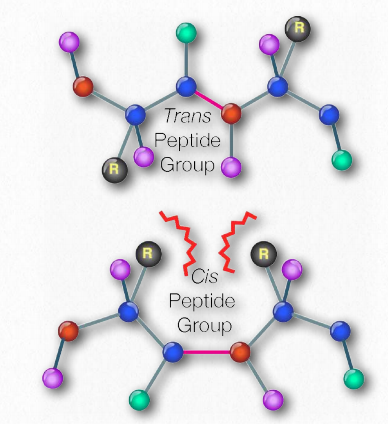
Esquemáticamente, en la Figura 2.18, podemos ver cómo los grupos R secuenciales de una proteína están dispuestos en una orientación alterna a cada lado de la cadena polipeptídica. La organización de los grupos R de esta manera no es aleatoria. El impedimento estérico puede ocurrir cuando los grupos R consecutivos están orientados en el mismo lado de una cadena principal peptídica (Figura 2.20)
Estructura primaria
La estructura primaria es el determinante final de la conformación general de una proteína. La estructura primaria de cualquier proteína llegó a su estado actual como resultado de la mutación y selección a lo largo del tiempo evolutivo. La estructura primaria de las proteínas es obligatoria por la secuencia de ADN que la codifica en el genoma. Las regiones de ADN que especifican proteínas se conocen como regiones codificantes (o genes).
Las secuencias de bases de estas regiones especifican directamente la secuencia de aminoácidos en las proteínas, con una correspondencia uno a uno entre los codones (grupos de tres bases consecutivas) en el ADN y los aminoácidos en la proteína codificada. La secuencia de codones en el ADN, copiados en ARN mensajero, especifica una secuencia de aminoácidos en una proteína. (Figura 2.21).
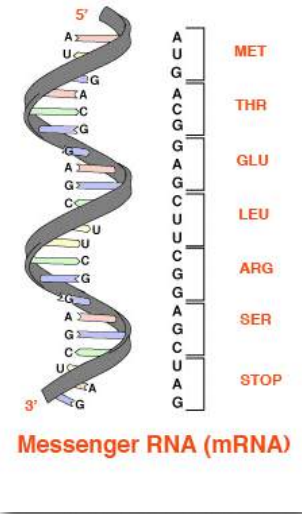
El orden en que los aminoácidos se unen en la síntesis de proteínas comienza a definir un conjunto de interacciones entre los aminoácidos incluso cuando la síntesis está ocurriendo. Es decir, un polipéptido puede plegarse incluso a medida que se está fabricando. El orden de las estructuras del grupo R y las interacciones resultantes son muy importantes porque las interacciones tempranas afectan las interacciones posteriores. Esto se debe a que las interacciones comienzan a establecer estructuras -secundarias y terciarias. Si una estructura helicoidal (estructura secundaria), por ejemplo, comienza a formarse, las posibilidades de interacción de un aminoácido particular Rgrupo pueden ser diferentes que si la hélice no se hubiera formado (Figura 2.22). Las interacciones del grupo R también pueden causar curvas en una secuencia polipeptídica (estructura terciaria) y estas curvas pueden crear (en algunos casos) oportunidades para interacciones que no habrían sido posibles sin la curva o prevenir (en otros casos) posibilidades de interacción similares.
Estructura secundaria
A medida que avanza la síntesis de proteínas, comienzan a ocurrir interacciones entre aminoácidos cercanos entre sí, dando lugar a patrones locales llamados estructura secundaria. Estas estructuras secundarias incluyen las conocidas hélices α y β-hebras. Ambos fueron predichos por Linus Pauling, Robert Corey y Herman Branson en 1951. Cada estructura tiene características únicas.
α-hélice
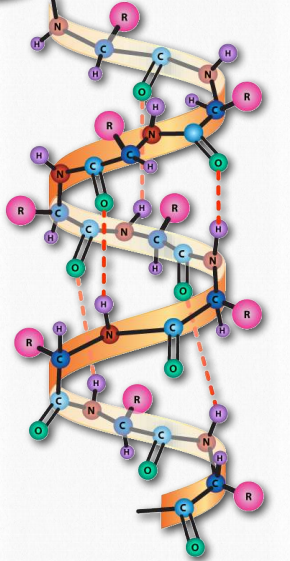
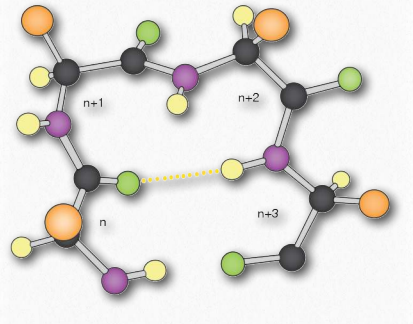
La hélice α tiene una estructura enrollada, con 3.6 aminoácidos por giro de la hélice (5 vueltas helicoidales = 18 aminoácidos). Las hélices son predominantemente diestras; solo en casos raros, como en secuencias con muchas glicinas se pueden formar hélices α- zurdas. En la hélice α, se forman enlaces de hidrógeno entre los grupos C=O y los grupos N-H en el esqueleto polipeptídico que están a cuatro aminoácidos distantes. Estos enlaces de hidrógeno son las fuerzas primarias que estabilizan la hélice α.
Usamos los términos rise, repeat y pitch para describir los parámetros de cualquier hélice. La repetición es el número de residuos en una hélice antes de que comience a repetirse. Para una hélice α, la repetición es de 3.6 aminoácidos por giro de la hélice. El ascenso es la distancia que la hélice eleva con la adición de cada residuo. Para una hélice α, esto es de 0.15 nm por aminoácido. El paso es la distancia entre giros completos de la hélice. Para una hélice α, esto es 0.54 nm. La estabilidad de una hélice α se ve potenciada por la presencia del aminoácido aspartato.

β hilo/hoja
Una hélice es, por supuesto, un objeto tridimensional. Una forma aplanada de hélice en dos dimensiones es una descripción común para una hebra β-. En lugar de bobinas, las hebras β tienen curvas y a veces se las conoce como pliegues, como los pliegues en una cortina. Los β-hilos se pueden organizar para formar estructuras elaboradamente organizadas, como láminas, barriles y otros arreglos.
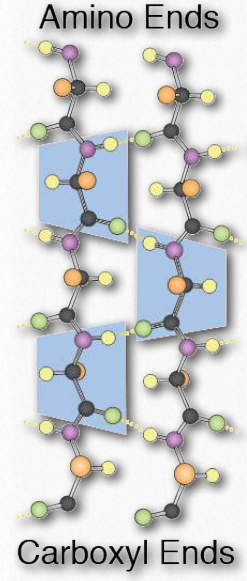
Las estructuras de cadena β de orden superior a veces se denominan estructuras supersecundarias), ya que implican interacciones entre aminoácidos no cercanas en secuencia primaria. Estas estructuras, también, se estabilizan mediante enlaces de hidrógeno entre átomos de oxígeno carbonilo e hidrógenos de grupos amina en la cadena principal polipeptídica (Figura 2.28). En una estructura de orden superior, las cadenas pueden disponerse paralelas (orientaciones amino a carboxilo iguales) o antiparalelas (orientaciones amino a carboxilo opuestas entre sí (en la Figura 2.27, la dirección de la hebra se muestra por la punta de flecha en los diagramas de cinta).
Giros
Los giros (a veces llamados giros inversos) son un tipo de estructura secundaria que, como su nombre indica, provoca un giro en la estructura de una cadena polipeptídica. Los giros dan lugar a la estructura terciaria en última instancia, provocando interrupciones en las estructuras secundarias (hélices α y β-hebras) y a menudo sirven como regiones de conexión entre dos regiones de estructura secundaria en una proteína. La prolina y la glicina juegan papeles comunes en los giros, proporcionando menos flexibilidad (comenzando el giro) y mayor flexibilidad (facilitando el giro), respectivamente.
Hay al menos cinco tipos de giros, con numerosas variaciones de cada uno dando lugar a muchos giros diferentes. Los cinco tipos de giros son
• δ-vueltas - los aminoácidos finales están separados por un enlace peptídico
• γ-vueltas - separación por dos enlaces peptídicos
•β-vueltas - separación por tres enlaces peptídicos
•α-vueltas - separación por cuatro enlaces peptídicos
•π-vueltas - separación por cinco enlaces
De estos, los giros β son la forma más común y los giros δ son teóricos, pero poco probables, debido a limitaciones estéricas. La Figura 2.29 representa un giro β-.
3 10 hélices
Además de la hélice α, las cadenas β y varios giros, se observan otras estructuras regulares que se repiten en las proteínas, pero ocurren con mucha menos frecuencia. La hélice 3 10 es la cuarta estructura secundaria más abundante en proteínas, constituyendo alrededor del 10-15% de todas las hélices. La hélice deriva su nombre del hecho de que contiene 10 aminoácidos en 3 vueltas. Es diestro. Se forman enlaces de hidrógeno entre los aminoácidos que están separados por tres residuos. Más comúnmente, la hélice 3 10 aparece en el extremo amino o carboxilo de una hélice α. Al igual que la hélice α, la hélice 3 10 se estabiliza por la presencia de aspartato en su secuencia.
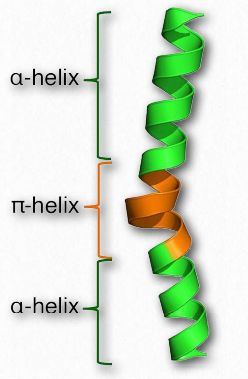
hélices π-
Una hélice π puede considerarse como un tipo especial de hélice α. Algunas fuentes lo describen como una hélice α con un aminoácido extra pegado en el medio de la misma (Figura 2.32). Las hélices π-no son exactamente raras, ocurriendo al menos una vez en tanto como 15% de todas las proteínas. Al igual que la hélice α, la hélice π-es diestra, pero donde la hélice α tiene 18 aminoácidos en 5 vueltas, la hélice π tiene 22 aminoácidos en 5 vueltas. Las hélices π-normalmente no se estiran por distancias muy largas. La mayoría tienen solo alrededor de 7 aminoácidos de largo y la secuencia casi siempre ocurre en el medio de una región α-helicoidal.
Parcelas Ramachandran
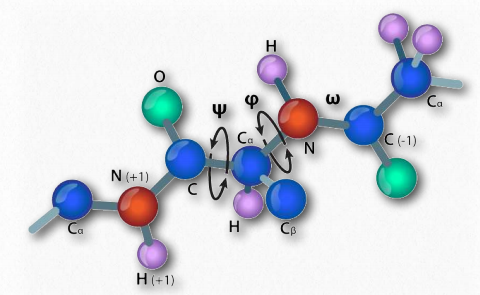
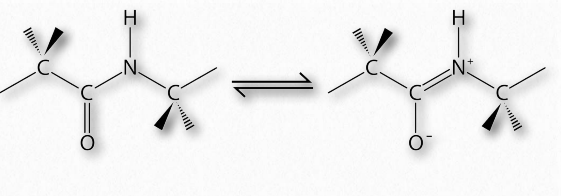
En 1963, G.N. Ramachandran, C. Ramakrishnan y V. Sasisekharan describieron una nueva forma de describir la estructura proteica. Si se considera la cadena principal de una cadena polipeptídica, consiste en un conjunto repetido de tres enlaces. Secuencialmente (en la dirección amino a carboxilo) son 1) un enlace giratorio (ψ) entre α-carbono y α-carboxilo que precede al enlace peptídico (ver AQUÍ), 2) un enlace peptídico no giratorio (ω) entre los grupos α-carboxilo y α-amina), y 3) un enlace giratorio (φ) entre la α-amina y α-carbono siguiendo el enlace peptídico (ver AQUÍ). Obsérvese en las Figuras 2.33 y 2.34 que la dirección amino a carboxilo es de derecha a izquierda.
La presencia del oxígeno carbonilo en el grupo α-carboxilo permite que el enlace peptídico exista como una estructura resonante, lo que significa que se comporta parte del tiempo como un doble enlace. Los dobles enlaces no pueden, por supuesto, rotar, pero los enlaces a ambos lados del mismo tienen cierta libertad de rotación. Los ángulos φ y ψ están restringidos a ciertos valores, ya que algunos ángulos darán como resultado un impedimento estérico. Además, cada tipo de estructura secundaria tiene un rango característico de valores para φ y ψ.
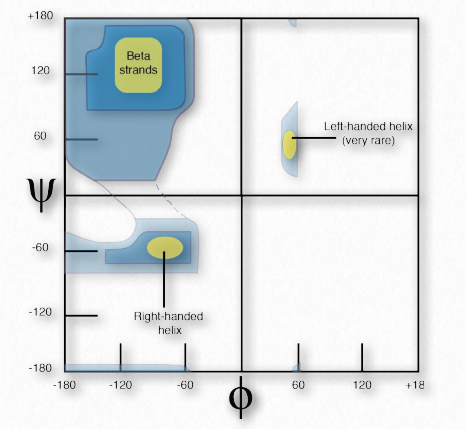
Ramachandran y sus colegas realizaron cálculos teóricos de la estabilidad energética de todos los ángulos posibles de 0° a 360° para cada uno de los ángulos φ y ψ y trazaron los resultados en una Gráfica Ramachandran (también llamada gráfica φ-ψ), delineando regiones de ángulos que teóricamente eran las más estables (Figura 2.35).
Se identificaron tres regiones primarias de estabilidad, correspondientes a ángulos φ-ψ de cadenas β (superior izquierda), hélices α- diestras (abajo izquierda) y hélices α-izquierdas (superior derecha). Las gráficas de estabilidad predicha son notablemente precisas cuando se comparan con los ángulos φ-ψ de las proteínas reales.
Predicción de la estructura secundaria
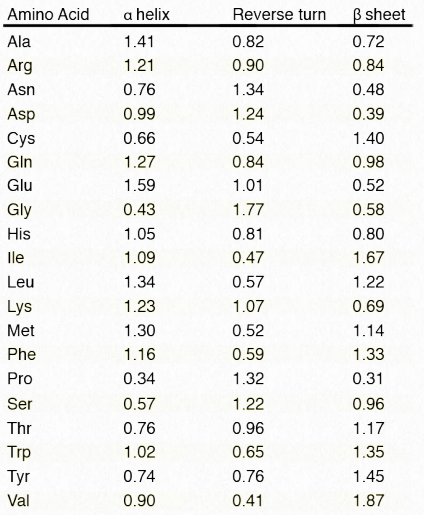
Cuadro 2.3 - Tendencias relativas de cada aminoácido a estar en una estructura secundaria. Valores más altos indican mayor tendencia Imagen por Penélope Irving
Al comparar la estructura primaria (secuencias de aminoácidos) con las estructuras conocidas de proteínas 3D, se puede contar cada vez que se encuentra un aminoácido en una hélice α, una fila/lámina β o un giro. El análisis por computadora de miles de estas secuencias permite asignar una probabilidad de que algún aminoácido dado aparezca en cada una de estas estructuras. Usando estas tendencias, uno puede, con una precisión de hasta un 80%, predecir regiones de estructura secundaria en una proteína basada únicamente en la secuencia de aminoácidos.
Esto se ve en el Cuadro 2.3. La ocurrencia en secuencia primaria de tres aminoácidos consecutivos con tendencias relativas superiores a uno es un indicador de que esa región del polipéptido se encuentra en la estructura secundaria correspondiente. Un recurso en línea para predecir estructuras secundarias llamado PSIPRED está disponible AQUÍ.
Hidrofobicidad
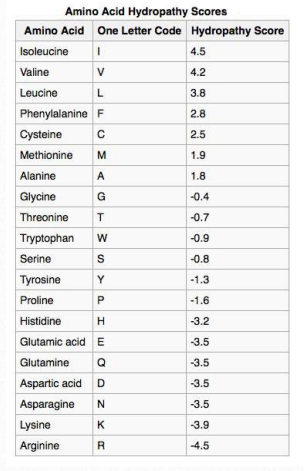
Cuadro 2.4 - Puntuaciones de hidropatía
La química de los grupos R de aminoácidos afecta las estructuras en las que se encuentran más comúnmente. Los subconjuntos de sus propiedades químicas pueden dar pistas sobre la estructura y, a veces, la ubicación celular. Un buen ejemplo es la hidrofobicidad (tendencias a evitar el agua) de algunos Rgrupos. Dado el ambiente acuoso de la célula, no es probable que tales grupos R estén en la superficie exterior de una proteína plegada.
Sin embargo, esta regla no es válida para regiones de proteína que puedan estar incrustadas dentro de las bicapas lipídicas de las membranas celulares/orgánulos. Esto se debe a que la región de tales proteínas que forman los dominios transmembrana están enterradas en el ambiente hidrófobo en el medio de la bicapa lipídica.
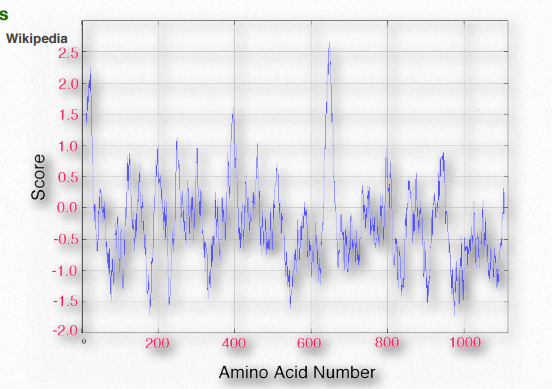
No es sorprendente que el escaneo de secuencias primarias para tramos específicamente dimensionados/espaciados de aminoácidos hidrófobos puede ayudar a identificar proteínas que se encuentran en las membranas. El Cuadro 2.4 muestra los valores de hidrofobicidad para los grupos R de los aminoácidos. En este conjunto, la escala va desde valores positivos (hidrófobos) hasta valores negativos (hidrófilos). En la Figura 2.36 se muestra una gráfica de hidropatía de KytedooLittle para la proteína de membrana del protooncogén RET. Dos regiones de la proteína son muy hidrofóbicas como se puede ver en los picos cerca de los aminoácidos 5-10 y 630-640. Se puede esperar razonablemente que tales regiones estén situadas dentro del interior de la proteína plegada o que formen parte de dominios transmembrana.
Bobinas aleatorias
Algunas secciones de una proteína no asumen una estructura regular y discernible y a veces se dice que carecen de estructura secundaria, aunque pueden tener enlaces de hidrógeno. Dichos segmentos se describen como que están en bobinas aleatorias y pueden tener fluidez en su estructura que da como resultado que tengan múltiples formas estables. Las bobinas aleatorias son identificables con métodos espectroscópicos, como el dicroísmo circular Wikipedia y la resonancia magnética nuclear (RMN) en la que se observan señales distintivas. Ver también proteínas metamórficas (AQUÍ) y proteínas intrínsecamente desordenadas (AQUÍ).

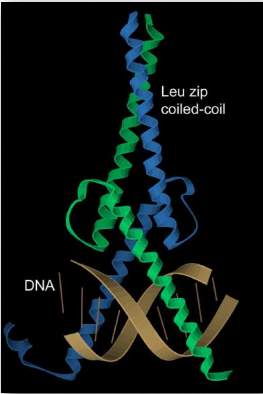
Estructura supersecundaria
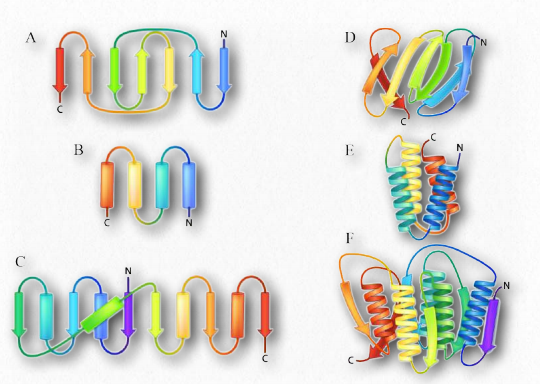
Otro elemento de la estructura proteica es más difícil de categorizar porque incorpora elementos de estructura secundaria y terciaria. Apodado estructura supersecundaria (o motivos estructurales), estas estructuras contienen múltiples componentes de estructura secundaria cercanos dispuestos de una manera específica y que aparecen en múltiples proteínas. Dado que hay muchas formas de hacer estructuras secundarias a partir de diferentes estructuras primarias, también pueden surgir motivos similares de diferentes secuencias primarias. Un ejemplo de un motivo estructural se muestra en la Figura 2.37.
Estructura terciaria
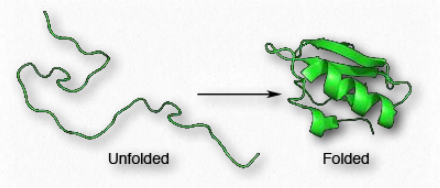
Las proteínas se distinguen entre sí por la secuencia de aminoácidos que las comprende. La secuencia de aminoácidos de una proteína determina la forma de la proteína, ya que las propiedades químicas de cada aminoácido son fuerzas que dan lugar a interacciones intermoleculares para comenzar a crear estructuras secundarias, como hélices α-y β-hebras. La secuencia también define giros y bobinas aleatorias que juegan un papel importante en el proceso de plegamiento de proteínas.
Dado que la forma es esencial para la función de las proteínas, la secuencia de aminoácidos da lugar a todas las propiedades que tiene una proteína. A medida que avanza la síntesis de proteínas, los componentes individuales de la estructura secundaria comienzan a interactuar entre sí, dando lugar a pliegues que acercan los aminoácidos que no están cerca uno del otro en la estructura primaria (Figura 2.38). A nivel terciario de estructura, las interacciones entre los grupos R de los aminoácidos en la proteína, así como entre la cadena principal del polipéptido y los grupos laterales de aminoácidos juegan un papel en el plegamiento.
Proteínas globulares
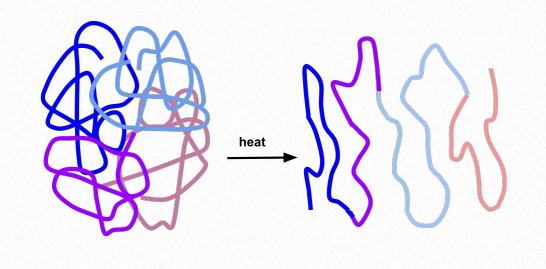
El plegamiento da lugar a distintas formas 3-D en proteínas que no son fibrosas. Estas proteínas se llaman globulares. Una proteína globular es estabilizada por las mismas fuerzas que impulsan su formación. Estos incluyen interacciones iónicas, enlaces de hidrógeno, fuerzas hidrófobas, enlaces iónicos, enlaces disulfuro y enlaces metálicos. Tratamientos como calor, cambios de pH, detergentes, urea y mercaptoetanol dominan las fuerzas estabilizadoras y provocan que una proteína se desdoble, perdiendo su estructura y (generalmente) su función (Figura 2.39). La capacidad del calor y los detergentes para desnaturalizar las proteínas es la razón por la que cocinamos nuestros alimentos y nos lavamos las manos antes de comer; dichos tratamientos desnaturalizan las proteínas en los microorganismos en nuestras manos. Los organismos que viven en ambientes de alta temperatura (más de 50°C) tienen proteínas con cambios en las fuerzas estabilizadoras: enlaces de hidrógeno adicionales, puentes salinos adicionales (interacciones iónicas) y compacidad pueden desempeñar un papel para evitar que estas proteínas se desplieguen.
Fuerzas estabilizadoras de proteínas
Antes de considerar el proceso de plegamiento, consideremos algunas de las fuerzas que ayudan a estabilizar las proteínas.
Enlaces de hidrógeno
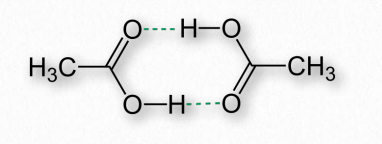
Los enlaces de hidrógeno surgen como resultado de los hidrógenos parcialmente cargados que se encuentran en los enlaces covalentes. Esto ocurre cuando el átomo al que está unido el hidrógeno tiene una electronegatividad mayor que el hidrógeno mismo, lo que resulta en que el hidrógeno tiene una carga positiva parcial porque no es capaz de retener electrones cerca de sí mismo (Figura 2.40).
El hidrógeno parcialmente cargado de esta manera es atraído por átomos, como el oxígeno y el nitrógeno que tienen cargas negativas parciales, debido a tener mayores electronegatividades y así mantener los electrones más cerca de sí mismos. Los hidrógenos parcialmente cargados positivamente se llaman donantes, mientras que los átomos parcialmente negativos a los que se sienten atraídos se llaman aceptores. (Ver Figura 1.30).
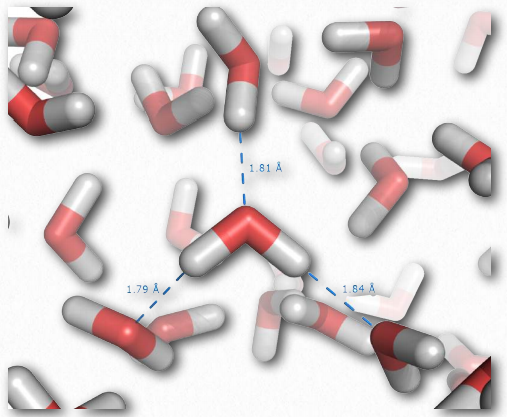
Los enlaces individuales de hidrógeno son mucho más débiles que un enlace covalente, pero colectivamente, pueden ejercer fuerzas fuertes. Considera el agua líquida, que contiene enormes cantidades de enlaces de hidrógeno (Figura 2.41). Estas fuerzas ayudan a que el agua permanezca líquida a temperatura ambiente. Otras moléculas que carecen de enlaces de hidrógeno de igual o mayor peso molecular que el agua, como el metano o el dióxido de carbono, son gases a la misma temperatura. Así, las interacciones intermoleculares entre las moléculas de agua ayudan a “mantener” el agua unida y seguir siendo un líquido. Notablemente, solo elevando la temperatura del agua a ebullición se superan las fuerzas de los enlaces de hidrógeno, permitiendo que el agua se vuelva completamente gaseosa.
Los enlaces de hidrógeno son fuerzas importantes en los biopolímeros que incluyen ADN, proteínas y celulosa. Todos estos polímeros pierden sus estructuras nativas al hervir. Los enlaces de hidrógeno entre aminoácidos que están cerca entre sí en estructura primaria pueden dar lugar a estructuras repetitivas regulares, como hélices o pliegues, en proteínas (estructura secundaria).
Interacciones iónicas
Las interacciones iónicas son fuerzas importantes que estabilizan la estructura proteica que surgen de la ionización de grupos R en los aminoácidos que comprenden una proteína. Estos incluyen los aminoácidos carboxilo (HERE), los aminoácidos amina así como el sulfhidrilo de la cisteína y a veces el hidroxilo de la tirosina.
Fuerzas hidrofóbicas
Las fuerzas hidrofóbicas estabilizan la estructura proteica como resultado de interacciones que favorecen la exclusión del agua. Los aminoácidos no polares (comúnmente encontrados en el interior de las proteínas) favorecen la asociación entre sí y esto tiene el efecto de excluir el agua. El agua excluida tiene una mayor entropía que el agua que interactúa con las cadenas laterales hidrofóbicas. Esto se debe a que el agua se alinea muy regularmente y en un patrón distinto al interactuar con moléculas hidrófobas.
Cuando se evita que el agua tenga este tipo de interacciones, está mucho más desordenada que lo estaría si pudiera asociarse con las regiones hidrofóbicas. Es en parte por esta razón que los aminoácidos hidrófobos se encuentran en los interiores de proteínas, por lo que pueden excluir el agua y aumentar la entropía.
Enlaces disulfuro
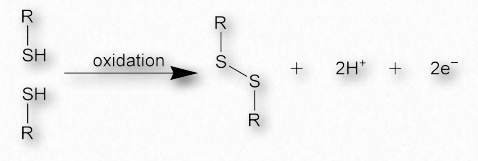
Los enlaces disulfuro, que se hacen cuando dos cadenas laterales sulfhidrilo de cisteína se acercan estrechamente, unen covalentemente diferentes regiones proteicas y pueden dar una gran fuerza a la estructura general (Figuras 2.42 y 2.43). Una oda a la estructura proteica por Kevin Ahern Los veinte aminos A's Definir una proteína de muchas maneras Su orden en una cadena peptídica Determina formas que ganan las proteínas Y cuando se enrollan, me deja alegre Cuz que hace estructuras secundarias Es terciario, me dicen Eso pasa cuando una proteína se pliega Pero cadenas plegadas son francamente aterradores Cuando se juntan cuaternario Son maravillas de la naturaleza, eso es seguro Creando problemas, haciendo curas Un tonto puede modelar poemas peptídicos Pero las proteínas provienen de ribosoems Estos residuos unidos de cisteína a veces se conocen como cistina. Los enlaces disulfuro son las fuerzas más fuertes que estabilizan la estructura de la proteína.

fuerzas van der Waals
fuerzas de van der Waals es un término utilizado para describir diversas interacciones débiles, incluidas las causadas por la atracción entre una molécula polar y un dipolo transitorio, o entre dos dipolos temporales. las fuerzas de van der Waals son dinámicas debido a la naturaleza fluctuante de la atracción, y generalmente son débiles en comparación con enlaces covalentes, pero puede, a distancias muy cortas, ser significativa.
Modificaciones postraduccionales
Las modificaciones postraduccionales también pueden dar como resultado la formación de enlaces covalentes que estabilizan proteínas. La hidroxilación de lisina y prolina en hebras de colágeno puede resultar en la reticulación de estos grupos y los enlaces covalentes resultantes ayudan a fortalecer y estabilizar el colágeno.
Modelos plegables
Actualmente se están investigando dos modelos populares de plegamiento de proteínas. En el primero (modelo de colisión por difusión), un evento de nucleación inicia el proceso, seguido de la formación de estructuras secundarias. Las colisiones entre las estructuras secundarias (como en la horquilla β en la Figura 2.37) permiten que se inicie el plegamiento. Por el contrario, en el modelo de nucleación-condensación, las estructuras secundaria y terciaria se forman juntas.
El plegamiento en las proteínas ocurre con bastante rapidez (0.1 a 1000 segundos) y puede ocurrir durante la síntesis; el extremo amino de una proteína puede comenzar a plegarse incluso antes de que se haga el extremo carboxilo, aunque ese no siempre es el caso.
Proceso de plegado
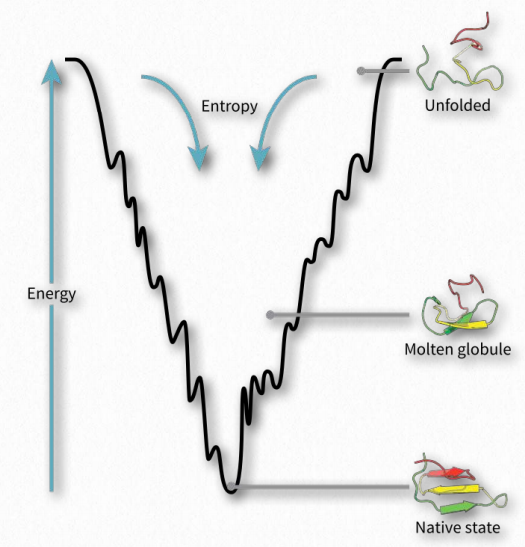
Se plantea la hipótesis de que el plegamiento de proteínas ocurre en un paisaje energético de “embudo plegable” en el que el estado nativo de una proteína plegada corresponde a la mínima energía libre posible en condiciones del medio (generalmente disolvente acuoso) en el que se disuelve la proteína. Como se ve en el diagrama (Figura 2.44), el embudo de energía tiene numerosos mínimos locales (inmersiones) en los que una proteína de plegamiento puede quedar atrapada a medida que se mueve hacia abajo de la parcela de energía. Otros factores, como la temperatura, los campos eléctricos/magnéticos y las consideraciones espaciales probablemente jueguen papeles.
Si las fuerzas externas afectan los mínimos de energía locales durante el plegado, el proceso y el producto final pueden ser influenciados. Como la velocidad de un automóvil que va por una carretera afectará la seguridad del viaje, también influyen las consideraciones energéticas y guían el proceso de plegado, lo que resulta en proteínas completamente funcionales, correctamente plegadas en algunos casos y “errores” mal plegados en otros.
Quedarse atascado
A medida que el proceso de plegado avanza hacia un mínimo de energía (fondo del embudo en la Figura 2.44), una proteína puede “atascarse” en cualquiera de los mínimos locales y no alcanzar el estado plegado final. Aunque el estado plegado es, en general, más organizado y por lo tanto ha reducido la entropía que el estado desplegado, hay dos fuerzas que superan la disminución de la entropía y impulsan el proceso hacia adelante.
El primero es la magnitud de la disminución de energía como se muestra en la gráfica. Dado que ΔG = ΔH -TΔS, una disminución en ΔH puede superar un ΔS negativo para hacer ΔG negativo y empujar el proceso de plegado hacia adelante. Las condiciones de energía favorables (disminuidas) surgen con la formación de enlaces iónicos, enlaces de hidrógeno, enlaces disulfuro y enlaces metálicos durante el proceso de plegado. Además, el efecto hidrofóbico aumenta la entropía al permitir que los aminoácidos hidrófobos en el interior de una proteína plegada excluyan el agua, contrarrestando así el impacto del orden de la estructura de la proteína al hacer que el ΔS sea menos negativo.
Predicción de estructura
Los programas de computadora son muy buenos para predecir la estructura secundaria basándose únicamente en la secuencia de aminoácidos, pero luchan con determinar la estructura terciaria usando la misma información. Esto se debe en parte al hecho de que las estructuras secundarias tienen puntos repetitivos de estabilización basados en la geometría y cualquier estructura secundaria regular (por ejemplo, α-hélice) varía muy poco de una a otra. Las estructuras plegadas, sin embargo, tienen un enorme número de estructuras posibles como lo muestra la Paradoja de Levinthal.
Espectroscopía
Debido a nuestra incapacidad para predecir con precisión la estructura terciaria basada en la secuencia de aminoácidos, las estructuras de las proteínas se determinan realmente usando técnicas de espectroscopia. En estos enfoques, las proteínas son sometidas a diversas formas de radiación electromagnética y las formas en que interactúan con la radiación permite a los investigadores determinar coordenadas atómicas a resolución Angstrom a partir de densidades de electrones (ver cristalografía de rayos X) y cómo interactúan los espines de los núcleos (ver RMN).
Paradoja de Levinthal
A finales de la década de 1960, Cyrus Levinthal describió la magnitud de la complejidad del problema del plegamiento de proteínas. Señaló que para una proteína con 100 aminoácidos, tendría 99 enlaces peptídicos y 198 consideraciones para ángulos φ y ψ. Si cada uno de estos tuviera sólo tres conformaciones, eso resultaría en 3198 plegados posibles diferentes o 2.95x1094.
Incluso permitiendo una cantidad razonable de tiempo (un nanosegundo) para que ocurra cada posible pliegue, tomaría más tiempo que la edad del universo muestrear todos ellos, lo que significa claramente que el proceso de plegamiento no se produce mediante un muestreo aleatorio secuencial y que intenta determinar la estructura de la proteína mediante los muestreos aleatorios fueron condenados al fracaso. Levinthal, por lo tanto, propuso que el plegamiento ocurre por un proceso secuencial que comienza con un evento de nucleación que guía el proceso rápidamente y no es diferente al proceso de embudo representado en la Figura 2.44.
Enfermedades del plegamiento erróneo de proteínas
El correcto plegamiento de las proteínas es esencial para su función. Se deduce entonces que el plegamiento erróneo de las proteínas (también llamado proteopatía) podría tener consecuencias. En algunos casos, esto podría simplemente resultar en una proteína inactiva. El mal plegamiento de proteínas también juega un papel en numerosas enfermedades, como la enfermedad de las vacas locas, la enfermedad de Alzheimer, la enfermedad de Parkinson y la enfermedad de Creutzfeldjakob. Muchas, pero no todas, las enfermedades de plegamiento erróneo afectan el tejido cerebral.
Depósitos insolubles
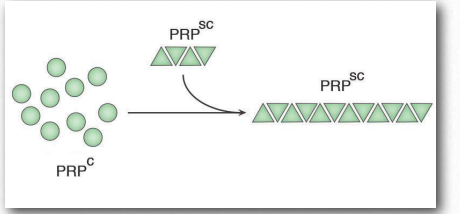
Las proteínas mal plegadas comúnmente formarán agregados llamados amiloides que son dañinos para los tejidos que las contienen porque cambian de ser solubles a insolubles en agua y forman depósitos. El proceso por el cual ocurre el plegamiento erróneo (Figura 2.45) no está completamente claro, pero en muchos casos, se ha demostrado que una proteína “semilla” que está mal plegada puede inducir el mismo plegamiento erróneo en otras copias de la misma proteína. Estas proteínas de semillas se conocen como priones y actúan como agentes infecciosos, resultando en la propagación de enfermedades. La lista de enfermedades humanas vinculadas al plegamiento erróneo de proteínas es larga y sigue creciendo. Un enlace de Wikipedia está AQUÍ.
Priones
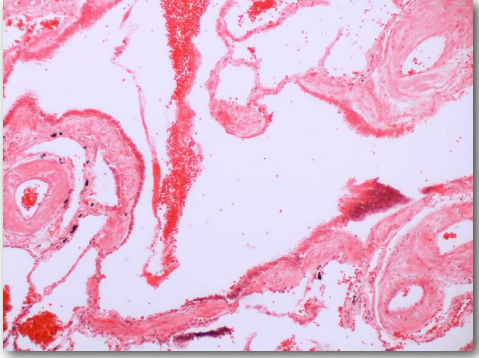
Los priones son partículas proteicas infecciosas que causan encefalopatías espongiformes transmisibles (EET), la más conocida de las cuales es la enfermedad de la Vaca Loca. Otras manifestaciones incluyen la enfermedad, tembladera, en ovejas y enfermedades humanas, como la enfermedad de Creutzfeldtjakob (ECJ), Insomnio familiar fatal y kuru. La proteína involucrada en estas enfermedades es una proteína de membrana llamada PrP. La PrP está codificada en el genoma de muchos organismos y se encuentra en la mayoría de las células del cuerpo. PrPC es el nombre que se le da a la estructura de PrP que es normal y no asociada a enfermedad. PrPSc es el nombre que se le da a una forma mal plegada de la misma proteína, que se asocia con el desarrollo de síntomas de la enfermedad (Figura 2.45).
Mal plegada
La PrPSc mal plegada se asocia con las enfermedades de EET y actúa como una partícula infecciosa. Una tercera forma de PrP, llamada PrPres, se puede encontrar en las EET, pero no es infecciosa. La 'res' de PrPres indica que es resistente a proteasas. Vale la pena señalar que las tres formas de PrP tienen la misma secuencia de aminoácidos y difieren entre sí solo en las formas en que se pliegan las cadenas polipeptídicas. La forma más peligrosamente mal plegada de PrP es PrPSc, debido a su capacidad para actuar como un agente infeccioso, una proteína de semilla que puede inducir el plegamiento erróneo de PrPc, convirtiéndola así en PrPSc.
Función
Se desconoce la función de PrPC. Los ratones que carecen del gen PrP no presentan anomalías mayores. Parece que presentan problemas con la memoria a largo plazo, lo que sugiere una función para PrPC. Stanley Prusiner, quien descubrió priones y acuñó el término, recibió el Premio Nobel de Medicina en 1997 por su trabajo. Creo que si por casualidad estuviera en Una proteína que compone un prión lo giraría y por el amor de Dios, dejaría de cometer errores de pliegue
Amiloides
Los amiloides son una colección de agregados proteicos plegados incorrectamente que se encuentran en el cuerpo humano. Como consecuencia de su mal plegamiento, son insolubles y contribuyen a una veintena de enfermedades humanas, incluidas importantes neurológicas que involucran priones. Las enfermedades incluyen (proteína afectada entre paréntesis) - enfermedad de Alzheimer (Amiloide β), enfermedad de Parkinson (α-sinucleína), enfermedad de Huntington (huntingtina), artritis reumatoide (amiloide sérico A), insomnio familiar fatal (PrPSc), y otras.
La secuencia de aminoácidos juega un papel en la amiloidogénesis. Los polipéptidos ricos en glutamina son comunes en levaduras y priones humanos. Las repeticiones de trinucleótidos son importantes en la enfermedad de Huntington. Donde la secuencia no es un factor, la asociación hidrofóbica entre las láminas β puede desempeñar un papel.
Amiloide β
Amiloide β se refiere a colecciones de pequeñas proteínas (36-43 aminoácidos) que parecen desempeñar un papel en la enfermedad de Alzheimer. (La proteína tau es el otro factor.) Son, de hecho, los principales componentes de las placas amiloides que se encuentran en los cerebros de pacientes que padecen la enfermedad y surgen de la escisión proteolítica de una glicoproteína precursora amiloide más grande llamada Proteína Precursora Amiloide, una proteína integral de membrana de células nerviosas cuya función no se conoce. Dos proteasas, β-secretasa y γ- secretasa realizan esta función. Las proteínas β amiloides están plegadas incorrectamente y parecen inducir a otras proteínas a plegarse erróneamente y así precipitar y formar la característica amiloide de la enfermedad. Las placas son tóxicas para las células nerviosas y dan lugar a la demencia característica de la enfermedad.
Se piensa que la agregación de proteínas β amiloides durante el plegamiento erróneo conduce a la generación de especies reactivas de oxígeno y que este es el medio por el cual se dañan las neuronas. No se sabe cuál es la función real del β amiloide. Las mutaciones autosómicas dominantes en la proteína conducen al inicio temprano de la enfermedad, pero esto ocurre en no más del 10% de los casos. Las estrategias para tratar la enfermedad incluyen la inhibición de las secretasas que generan los fragmentos peptídicos a partir de la proteína precursora amiloide.
Huntingtina
La huntingtina es el gen central de la enfermedad de Huntington. La proteína que se elabora a partir de ella es rica en glutamina, con 6-35 de tales residuos en su forma de tipo silvestre. En la enfermedad de Huntington, este gen está mutado, aumentando el número de glutaminas en la proteína mutante a entre 36 y 250. El tamaño de la proteína varía con el número de glutaminas en la proteína mutante, pero la proteína de tipo salvaje tiene más de 3100 aminoácidos y un peso molecular de aproximadamente 350,000 Da. Se desconoce su función precisa, pero la huntingtina se encuentra en las células nerviosas, con el nivel más alto en el cerebro. Se cree que posiblemente desempeñe papeles en el transporte, la señalización y la protección contra la apoptosis. La huntingtina también es necesaria para el desarrollo embrionario temprano. Dentro de la célula, la huntingtina se encuentra localizada principalmente con microtúbulos y vesículas.
Repetición de trinucleótidos
El gen de la huntingtina contiene muchas copias de la secuencia CAG (llamadas repeticiones de trinucleótidos), que codifican para las muchas glutaminas en la proteína. La enfermedad de Huntington surge cuando se generan copias adicionales de la secuencia CAG cuando se está copiando el ADN del gen. La expansión de secuencias repetidas puede ocurrir debido al deslizamiento de la polimerasa con respecto al molde de ADN durante la replicación. Como resultado, se pueden hacer múltiples copias adicionales de la repetición del trinucleótido, dando como resultado proteínas con números variables de residuos de glutamina. Se pueden tolerar hasta 35 repeticiones sin problema. El número de repeticiones puede expandirse a lo largo de la vida de una persona, sin embargo, por el mismo mecanismo. Los individuos con 36-40 repeticiones comienzan a mostrar signos de la enfermedad y si hay más de 40, la enfermedad estará presente.
Chaperonas moleculares
La importancia del correcto plegamiento de las proteínas se destaca por las enfermedades asociadas a las proteínas mal plegadas, por lo que no es de extrañar, entonces, que las células gasten energía para facilitar el correcto plegamiento de las proteínas. Las células utilizan dos clases de proteínas conocidas como chaperonas moleculares, para facilitar dicho plegamiento en las células. Las chaperonas moleculares son de dos tipos, las chaperonas y las chaperoninas. Un ejemplo de la primera categoría es la clase de proteínas Hsp70. Hsp significa “proteína de choque térmico”, con base en que estas proteínas se observaron por primera vez en grandes cantidades en células que habían sido sometidas brevemente a altas temperaturas. Las Hsps funcionan para ayudar a las células en tensiones derivadas del choque térmico y la exposición a condiciones oxidantes o metales pesados tóxicos, como el cadmio y el mercurio. Sin embargo, también juegan un papel importante en condiciones normales, donde ayudan en el plegamiento adecuado de los polipéptidos al prevenir interacciones aberrantes que podrían conducir a un plegamiento incorrecto o agregación. Las proteínas Hsp70 se encuentran en casi todas las células y utilizan la hidrólisis de ATP para estimular cambios estructurales en la forma de la chaperona para acomodar la unión de proteínas sustrato. El dominio de unión de HSP70s contiene una estructura β-barril que se envuelve alrededor de la cadena polipeptídica del sustrato y tiene afinidad por cadenas laterales hidrófobas de aminoácidos. Como se muestra en la Figura 2.50, la Hsp70 se une a polipéptidos a medida que emergen de los ribosomas durante la síntesis de proteínas. La unión del sustrato estimula la hidrólisis de ATP y esto se ve facilitado por otra proteína de choque térmico conocida como Hsp40. La hidrólisis de ATP hace que la Hsp70 tome una conformación cerrada que ayuda a proteger los residuos hidrófobos expuestos y evitar la agregación o el plegamiento local.
Una vez completada la síntesis de proteínas, el ADP se libera y se reemplaza por ATP y esto da como resultado la liberación de la proteína sustrato, lo que luego permite que el polipéptido de longitud completa se pliegue correctamente.
En choque térmico
En tiempos de choque térmico o estrés oxidativo, las proteínas Hsp70 se unen a regiones hidrofóbicas desplegadas de proteínas para evitar de manera similar que se agreguen y permitan que se replegen adecuadamente. Cuando las proteínas están dañadas, Hsp70 recluta enzimas que ubiquitinan la proteína dañada para atacarlas para su destrucción en proteasomas. Por lo tanto, las proteínas Hsp70 juegan un papel importante para garantizar no solo que las proteínas se plieguen adecuadamente, sino que las proteínas dañadas o no funcionales se eliminen por degradación en el proteasoma.
Chaperoninas
Una segunda clase de proteínas involucradas en ayudar a otras proteínas a plegarse adecuadamente se conocen como chaperoninas. Hay dos categorías primarias de chaperoninas: Clase I (encontrada en bacterias, cloroplastos y mitocondrias) y Clase II (encontrada en el citosol de eucariotas y arquebacterias). Las chaperoninas mejor estudiadas son las proteínas del complejo Groel/Groes que se encuentran en bacterias (Figura 2.51).
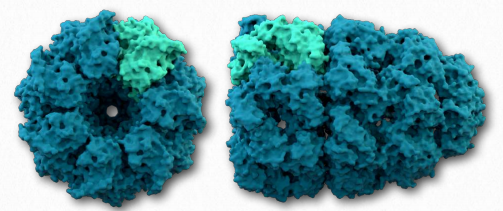
Los groes/groes pueden no ser capaces de deshacer las proteínas agregadas, pero al facilitar el plegamiento adecuado, proporciona competencia por el plegamiento incorrecto como proceso y puede reducir o eliminar los problemas que surgen del plegamiento inadecuado. GroEl es un 14mero de doble anillo con una región hidrófoba que puede facilitar el plegamiento de sustratos de 15-60 kDa de tamaño. GroES es un heptámero singlering que se une a GroEl en presencia de ATP y funciona como una cubierta sobre GroEl. La hidrólisis de ATP por chaperoninas induce grandes cambios conformacionales que afectan la unión de proteínas sustrato y su plegamiento. No se sabe exactamente cómo las chaperoninas pliegan las proteínas. Los modelos pasivos postulan que el complejo chaperonina funciona de forma inercia al evitar interacciones intermoleculares desfavorables o al poner restricciones en los espacios disponibles para que se produzca el plegado. Los modelos activos proponen que los cambios estructurales en el complejo chaperonina inducen cambios estructurales en la proteína sustrato.
Desglose de proteínas
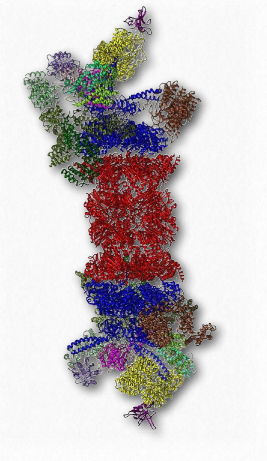
Otro complejo proteico que tiene una función importante en la dinámica de vida de las proteínas es el proteasoma (Figura 2.52). Los proteasomas, que se encuentran en todos los eucariotas y arqueos, así como algunas bacterias, funcionan para descomponer proteínas innecesarias o dañadas por degradación proteolítica. Los proteasomas ayudan a regular la concentración de algunas proteínas y degradan las que están mal plegadas. La vía de degradación proteasómica juega un papel importante en los procesos celulares que incluyen la progresión a través del ciclo celular, la modulación de la expresión génica y la respuesta al estrés oxidativo.
La degradación en el proteasoma produce péptidos cortos de siete a ocho aminoácidos de longitud. Las treoninas proteasas juegan un papel importante. La descomposición de estos péptidos produce aminoácidos individuales, facilitando así su reciclaje en las células. Las proteínas se dirigen a la degradación en proteasomas eucariotas mediante la unión a múltiples copias de una pequeña proteína llamada ubiquitina (8.5 kDa - 76 aminoácidos). La enzima que cataliza la reacción se conoce como ubiquitina ligasa. La cadena de poliubiquitina resultante está unida por el proteasoma y comienza la degradación. La ubiquitina fue nombrada debido a que se encuentra ubicuamente en células eucariotas.
Ubiquitina
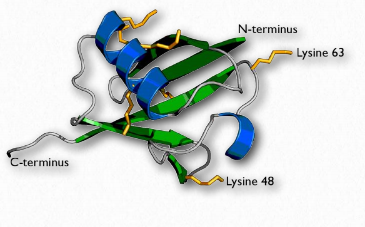
La ubiquitina (Figura 2.53) es una proteína multifuncional pequeña (8.5 kDa) que se encuentra en células eucariotas. Comúnmente se agrega a las proteínas diana por la acción de las enzimas ubiquitina ligasa (E3 en la Figura 2.54). Se pueden agregar una (ubiquitinación) o muchas (poliubiquitinación) moléculas de ubiquitina. La unión de la ubiquitina es a través de la cadena lateral de uno de los siete residuos de lisina diferentes en ubiquitina.
La adición de ubiquitina a las proteínas tiene muchos efectos, el más conocido de los cuales es apuntar a la proteína para su degradación en el proteasoma. La diana proteasomal se observa cuando la poliubiquitinación ocurre en las lisinas #29 y 48. La poliubiquitinación o monoubiquitinación en otras lisinas puede resultar en una localización celular alterada y en interacciones proteína-proteína cambiadas. Estos últimos pueden alterar la inflamación, el tráfico endocítico, la traducción y la reparación del ADN.
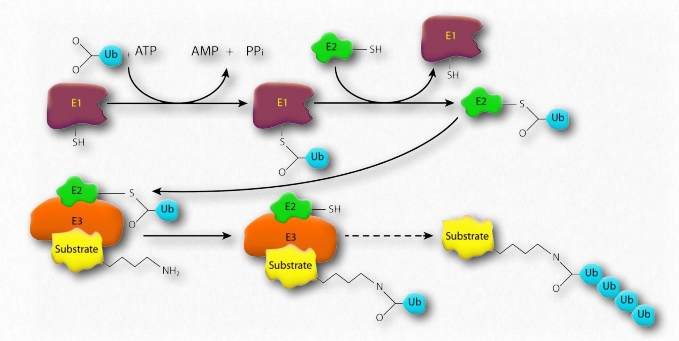
Mal funcionamiento de la ubiquitina ligasa
Parkin es una proteína relacionada con la enfermedad de Parkinson que, cuando muta, se vincula a una forma heredada de la enfermedad llamada enfermedad de Parkinson juvenil autosómica recesiva. Se desconoce la función de la proteína, pero es un componente del sistema E3 ubiquitina ligasa responsable de transferir ubiquitina de la proteína E2 a una cadena lateral de lisina en la proteína diana. Se piensa que las mutaciones en parkin conducen a una disfunción proteasomal y una consecuente incapacidad para descomponer proteínas dañinas para las neuronas dopaminérgicas. Esto da como resultado la muerte o mal funcionamiento de estas neuronas, resultando en la enfermedad de Parkinson.
Proteínas intrínsecamente desordenadas
Película 2.1 - Movimiento dinámico del citocromo C en solución Wikipedia
Como es evidente a partir de los muchos ejemplos descritos en otra parte del libro, la estructura 3-D de las proteínas es importante para su función. Pero, cada vez más, se hace evidente que no todas las proteínas se pliegan en una estructura estable. Estudios sobre las llamadas proteínas intrínsecamente desordenadas (IDP) en las últimas décadas han demostrado que muchas proteínas son biológicamente activas, incluso pensadas que no se pliegan en estructuras estables. Aún otras proteínas exhiben regiones que permanecen desplegadas (regiones IDP) incluso cuando el resto del polipéptido se pliega en una forma estructurada.
Las proteínas intrínsecamente desordenadas y las regiones desordenadas dentro de las proteínas, de hecho, se conocen desde hace muchos años, pero fueron consideradas como una anomalía. Es solo recientemente, al darse cuenta de que los IDP y las regiones IDP están muy extendidas entre las proteínas eucariotas, que se ha reconocido que el trastorno observado es una “característica, no un error”.
Película 2.2 SUMO-1, una proteína con intrínsecamente desordenado secciones Wikipedia
La comparación de los IDP muestra que comparten características de secuencia que parecen favorecer su estado desordenado. Es decir, así como algunas secuencias de aminoácidos pueden favorecer el plegamiento de un polipéptido en una estructura particular, las secuencias de aminoácidos de los IDP favorecen su resto desplegado. Se observa que las regiones IDP son bajas en residuos hidrófobos e inusualmente ricas en residuos polares y prolina. La presencia de un gran número de aminoácidos cargados en los IDP puede inhibir el plegamiento a través de la repulsión de carga, mientras que la falta de residuos hidrófobos dificulta la formación de un núcleo hidrófobo estable, y la prolina desalienta la formación de estructuras helicoidales. Las diferencias observadas entre secuencias de aminoácidos en IDP y proteínas estructuradas se han utilizado para diseñar algoritmos para predecir si una secuencia de aminoácidos dada estará desordenada.
¿Cuál es el significado de las proteínas o regiones intrínsecamente desordenadas? El hecho de que esta propiedad esté codificada en sus secuencias de aminoácidos sugiere que su trastorno puede estar vinculado a su función. La naturaleza flexible y móvil de algunas regiones IDP puede desempeñar un papel crucial en su función, permitiendo una transición a una estructura plegada al unirse a una pareja proteica o someterse a una modificación postraduccional. Estudios sobre varias proteínas bien conocidas con regiones IDP sugieren algunas respuestas. Las regiones IDP pueden mejorar la capacidad de proteínas como el represor lac para translocarse a lo largo del ADN para buscar sitios de unión específicos. La flexibilidad de los IDP también puede ser un activo en las interacciones proteína-proteína, especialmente para las proteínas que se sabe que interactúan con muchos socios proteicos diferentes.
Por ejemplo, p53 tiene regiones IDP que pueden permitir que la proteína interactúe con una variedad de parejas funcionales. La comparación de las funciones conocidas de las proteínas con predicciones de trastorno en estas proteínas sugiere que las regiones IDP y IDP pueden funcionar desproporcionadamente en la señalización y regulación, mientras que las proteínas más estructuradas se inclinan hacia roles en la catálisis y el transporte. Curiosamente, se predice que muchas de las proteínas que se encuentran tanto en los ribosomas como en los empalmeosomas tienen regiones IDP que pueden desempeñar un papel en el ensamblaje correcto de estos complejos. A pesar de que los IDP no han sido estudiados intensamente desde hace mucho tiempo, lo poco que se sabe de ellos sugiere que desempeñan un papel importante y subestimado en las células.
Proteínas metamórficas
Otro grupo de proteínas que recientemente han cambiado nuestro pensamiento sobre la estructura y función de las proteínas son las llamadas proteínas metamórficas. Estas proteínas son capaces de formar más de un estado estable, plegado comenzando con una sola secuencia de aminoácidos. Si bien es cierto que las conformaciones plegadas múltiples no están descartadas por las leyes de la física y la química, las proteínas metamórficas son un descubrimiento relativamente nuevo. Se sabía, por supuesto, que las proteínas priónicas eran capaces de plegarse en estructuras alternativas, pero las proteínas metamórficas parecen ser capaces de alternar de un lado a otro entre dos estructuras estables. Mientras que en algunos casos, la proteína metamórfica sufre este cambio en respuesta a unirse a otra molécula, algunas proteínas que pueden lograr esta transición por sí mismas. Un ejemplo interesante es la molécula de señalización, la linfoactina. La linfoactina tiene dos funciones biológicas que se llevan a cabo por sus dos confórmeros: una forma monomérica que se une al receptor de linfoactina y una forma dimérica que se une a la heparina. Es posible que este tipo de conmutación esté más extendido de lo que se ha pensado.
Replegamiento de proteínas desnaturalizadas
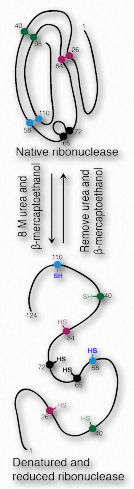
Toda la información para el plegamiento de proteínas está contenida en la secuencia de aminoácidos de la proteína. Puede parecer curioso entonces que la mayoría de las proteínas no se plieguen en su forma adecuada y completamente activa después de que hayan sido desnaturalizadas +++ y se elimine el desnaturalizante. Algunos lo hacen, de hecho. Un buen ejemplo es la ribonucleasa bovina (Figura 2.55). Su actividad catalítica es muy resistente al calor y a la urea y los intentos de desnaturalizarla no funcionan muy bien. Sin embargo, si se trata la enzima con β-mercaptoetanol (que rompe los enlaces disulfuro) antes del tratamiento con urea y/o calentamiento, se pierde actividad, lo que indica que los enlaces disulfuro covalentes ayudan a estabilizar la estructura general de la enzima y cuando se rompen, la desnaturalización puede ocurrir fácilmente. Cuando la mezcla se enfría de nuevo a temperatura ambiente, con el tiempo reaparece alguna actividad enzimática, lo que indica que la ribonucleasa se volvió a plegar bajo las nuevas condiciones.
Curiosamente, la renaturalización ocurrirá al máximo si se deja una pequeña cantidad de β-mercaptoetanol en la solución durante el proceso. La razón de esto es porque el β- mercaptoetanol permite la reducción (y ruptura) de enlaces disulfuro accidentales e incorrectos durante el proceso de plegamiento. Sin él, estos enlaces disulfuro evitarán que se formen pliegues adecuados.
Desdesnaturalización irreversible
La mayoría de las enzimas, sin embargo, no se comportan como ribonucleasa bovina. Una vez desnaturalizada, su actividad no se puede recuperar a ningún significativo No hay muchas maneras Inactivar RNasa Es estable cuando hace calor o frío Porque los disulfuros se mantienen firmemente Si deseas que se detenga Usa extensión de mercaptoetanol caliente. Esto puede parecer contradecir la idea de que la información de plegamiento es inherente a la secuencia de aminoácidos en la proteína. No lo hace.
La mayoría de las enzimas no se replegan correctamente después de la desnaturalización por dos razones. En primer lugar, el plegamiento normal puede ocurrir a medida que se hacen las proteínas. Las interacciones entre aminoácidos tempranos en la síntesis no se “confunden” por interacciones con aminoácidos posteriores en la síntesis porque esos aminoácidos no están presentes cuando comienza el proceso.
El papel de los chaperoninos
En otros casos, el proceso de plegamiento de algunas proteínas en la célula se basó en la acción de las proteínas chaperoninas (ver AQUÍ). En ausencia de chaperoninas, ocurren interacciones que podrían resultar en plegamiento erróneo, evitando así el correcto plegamiento. Así, el plegamiento temprano y la asistencia de las chaperoninas eliminan algunas interacciones potenciales de “plegamiento erróneo” que pueden ocurrir si toda la secuencia estaba presente cuando comenzó el plegamiento.
Estructura cuaternaria
Un cuarto nivel de estructura proteica es el de la estructura cuaternaria. Se refiere a estructuras que surgen como resultado de interacciones entre múltiples polipéptidos. Las unidades pueden ser múltiples copias idénticas o pueden ser diferentes cadenas polipeptídicas. La hemoglobina adulta es un buen ejemplo de una proteína con estructura cuaternaria, estando compuesta por dos cadenas idénticas llamadas α y dos cadenas idénticas llamadas β.
Aunque las cadenas α son muy similares a las cadenas β, no son idénticas. Tanto las cadenas α como las β-cadenas también están relacionadas con la cadena polipeptídica única en la proteína relacionada llamada mioglobina. Tanto la mioglobina como la hemoglobina tienen similitud en la unión del oxígeno, pero su comportamiento hacia la molécula difiere significativamente. Notablemente, las múltiples subunidades de hemoglobina (con estructura cuaternaria) en comparación con la subunidad única de mioglobina (sin estructura cuaternaria) dan lugar a estas diferencias.
Referencias
1. https://en.wikipedia.org/wiki/Van_der_W aals_force 105