
14.3: Medicamentos dirigidos a otros microorganismos
- Page ID
- 54916

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Objetivos de aprendizaje
- Describir los mecanismos de acción asociados con fármacos que inhiben la biosíntesis de la pared celular, la síntesis de proteínas, la función de membrana, la síntesis de ácidos nucleicos y la vía metabólica.
Una cualidad importante para un fármaco antimicrobiano es la toxicidad selectiva, lo que significa que mata o inhibe selectivamente el crecimiento de dianas microbianas al tiempo que causa un daño mínimo o nulo al huésped. La mayoría de los medicamentos antimicrobianos actualmente en uso clínico son antibacterianos porque la célula procariota proporciona una mayor variedad de dianas únicas para toxicidad selectiva, en comparación con hongos, parásitos y virus. Cada clase de fármacos antibacterianos tiene un modo de acción único (la forma en que un fármaco afecta a los microbios a nivel celular), y estos se resumen en Figura\(\PageIndex{1}\) y Tabla\(\PageIndex{1}\).
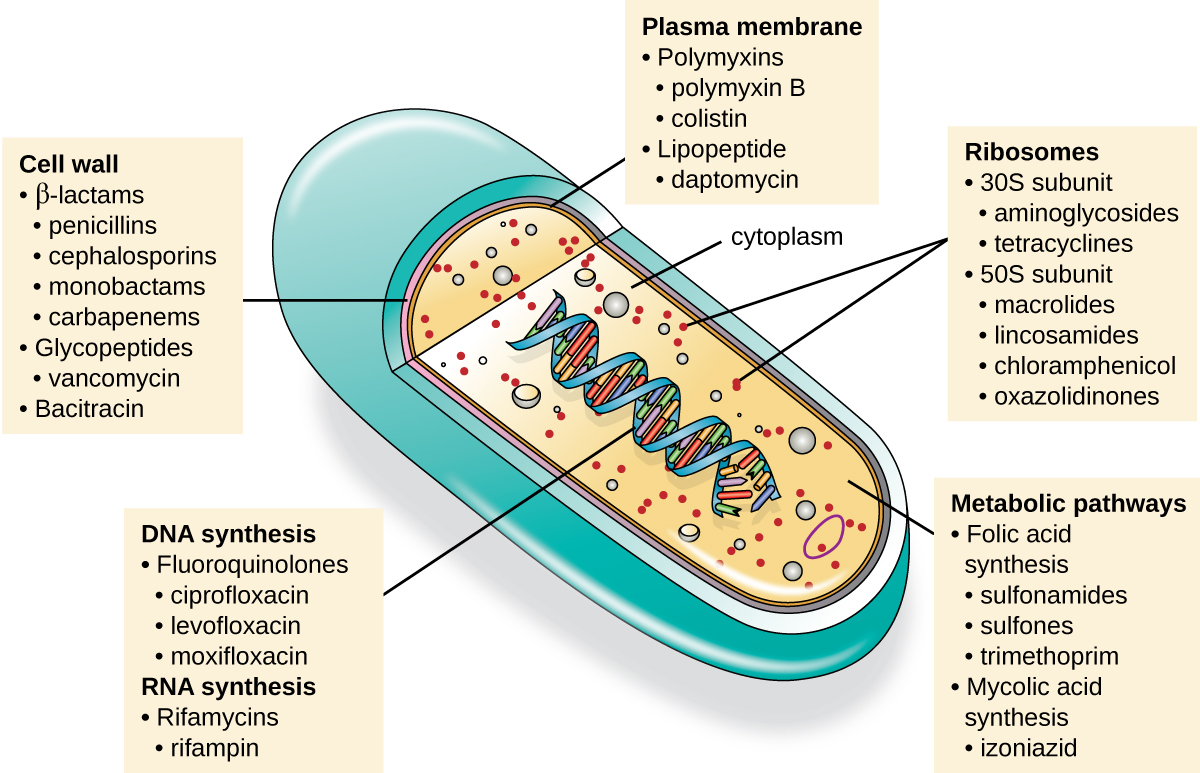
| Modo de Acción | Target | Clase de medicamento |
|---|---|---|
| Inhibir la biosíntesis de pared celular | Proteínas de unión a penicilina | β-lactamas: penicilinas, cefalosporinas, monobactámicos, carbapenémicos |
| Subunidades de peptidoglicano | Glucopéptidos | |
| Transporte de la subunidad de peptidoglicano | Bacitracina | |
| Inhibir la biosíntesis de proteínas | Subunidad ribosómica 30S | Aminoglucósidos, tetraciclinas |
| Subunidad ribosómica 50S | Macrólidos, lincosamidas, cloranfenicol, oxazolidinonas | |
| Interrumpen membranas | Lipopolisacárido, membranas internas y externas | Polimixina B, colistina, daptomicina |
| Inhibir síntesis de ácidos nucleicos | RNA | Rifamicina |
| ADN | Fluoroquinolonas | |
| Antimetabolitos | Enzima de síntesis de ácido | sulfonamidas, trimetoprima |
| Enzima de síntesis de ácido micólico | Hidrazida de ácido isonicotínico | |
| Inhibidor micobacteriano de la adenosina trifosfato (ATP) sintasa | ATP sintasa micobacteriana | Diarilquinolina |
Inhibidores de la biosíntesis de pared celular
Varias clases diferentes de antibacterianos bloquean los pasos en la biosíntesis del peptidoglicano, haciendo que las células sean más susceptibles a la lisis osmótica (Tabla\(\PageIndex{2}\)). Por lo tanto, los antibacterianos que se dirigen a la biosíntesis de la pared celular son bactericidas en su acción. Debido a que las células humanas no producen peptidoglicano, este modo de acción es un excelente ejemplo de toxicidad selectiva.
La penicilina, el primer antibiótico descubierto, es uno de varios antibacterianos dentro de una clase llamada β-lactamas. Este grupo de compuestos incluye las penicilinas, cefalosporinas, monobactamas y carbapenémicos, y se caracteriza por la presencia de un anillo β-lactama que se encuentra dentro de la estructura central de la molécula de fármaco (Figura\(\PageIndex{2}\)). Los antibacterianos β-lactámicos bloquean la reticulación de cadenas peptídicas durante la biosíntesis de nuevo peptidoglicano en la pared celular bacteriana. Son capaces de bloquear este proceso porque la estructura β-lactama es similar a la estructura del componente de la subunidad peptidoglicano que es reconocida por la enzima transpeptidasa reticulante, también conocida como proteína de unión a penicilina (PBP). Aunque el anillo β-lactama debe permanecer inalterado para que estos fármacos conserven su actividad antibacteriana, los cambios químicos estratégicos en los grupos R han permitido el desarrollo de una amplia variedad de fármacos β-lactámicos semisintéticos con mayor potencia, espectro de actividad ampliado y vidas medias más largas para mejorar dosificación, entre otras características.
La penicilina G y la penicilina V son antibióticos naturales de hongos y son principalmente activos contra patógenos bacterianos grampositivos, y algunos patógenos bacterianos gramnegativos como Pasteurella multocida. La figura\(\PageIndex{2}\) resume el desarrollo semisintético de algunas de las penicilinas. Al agregar un grupo amino (-NH 2) a la penicilina G se crearon las aminopenicilinas (es decir, ampicilina y amoxicilina) que han aumentado el espectro de actividad contra más patógenos gramnegativos. Además, la adición de un grupo hidroxilo (-OH) a la amoxicilina incrementó la estabilidad ácida, lo que permite una mejor absorción oral. La meticilina es una penicilina semisintética que se desarrolló para abordar la propagación de enzimas (penicilinasas) que estaban inactivando las otras penicilinas. Cambiar el grupo R de penicilina G por el grupo dimetoxifenilo más voluminoso proporcionó protección del anillo β-lactama de la destrucción enzimática por penicilinasas, dándonos la primera penicilina resistente a penicilinasas.
Similar a las penicilinas, las cefalosporinas contienen un anillo β-lactama (Figura\(\PageIndex{2}\)) y bloquean la actividad transpeptidasa de las proteínas de unión a penicilina. Sin embargo, el anillo β-lactama de las cefalosporinas se fusiona a un anillo de seis miembros, en lugar del anillo de cinco miembros que se encuentra en las penicilinas. Esta diferencia química proporciona a las cefalosporinas una mayor resistencia a la inactivación enzimática por β-lactamasas. El fármaco cefalosporina C se aisló originalmente del hongo Cephalosporium acremonium en la década de 1950 y tiene un espectro de actividad similar al de la penicilina contra bacterias grampositivas pero es activo contra más bacterias gramnegativas que la penicilina. Otra diferencia estructural importante es que la cefalosporina C posee dos grupos R, en comparación con un solo grupo R para penicilina, lo que proporciona una mayor diversidad en alteraciones químicas y desarrollo de cefalosporinas semisintéticas. La familia de cefalosporinas semisintéticas es mucho mayor que las penicilinas, y estos fármacos se han clasificado en generaciones basándose principalmente en su espectro de actividad, aumentando en espectro desde las cefalosporinas de primera generación de espectro estrecho hasta las de amplio espectro, cuarta generación cefalosporinas. Se ha desarrollado una nueva cefalosporina de quinta generación que es activa contra Staphylococcus aureus resistente a meticilina (SARM).
Los carbapenémicos y monobactámicos también tienen un anillo β-lactama como parte de su estructura central, e inhiben la actividad transpeptidasa de las proteínas de unión a penicilina. El único monobactam utilizado clínicamente es el aztreonam. Es un antibacteriano de espectro estrecho con actividad solo contra bacterias gramnegativas. En contraste, la familia de carbapenem incluye una variedad de fármacos semisintéticos (imipenem, meropenem y doripenem) que proporcionan actividad de muy amplio espectro contra patógenos bacterianos grampositivos y gramnegativos.
El fármaco vancomicina, miembro de una clase de compuestos llamados glicopéptidos, fue descubierto en la década de 1950 como un antibiótico natural del actinomiceto Amycolatopsis orientalis. Similar a las β-lactamas, la vancomicina inhibe la biosíntesis de la pared celular y es bactericida. Sin embargo, en contraste con las β-lactamas, la estructura de la vancomicina no es similar a la de las subunidades de peptidoglicano de la pared celular y no inactiva directamente las proteínas de unión a penicilina. Más bien, la vancomicina es una molécula muy grande y compleja que se une al extremo de la cadena peptídica de los precursores de la pared celular, creando un bloqueo estructural que evita que las subunidades de la pared celular se incorporen en la cadena principal de N-acetilglucosamina y ácido N-acetilmurámico (NAM-NAG) en crecimiento del peptidoglicano estructura (transglicosilación). La vancomicina también bloquea estructuralmente la transpeptidación. La vancomicina es bactericida contra patógenos bacterianos grampositivos, pero no es activa contra bacterias gramnegativas debido a su incapacidad para penetrar en la membrana externa protectora.
El fármaco bacitracina consiste en un grupo de antibióticos peptídicos estructuralmente similares originalmente aislados de Bacillus subtilis. La bacitracina bloquea la actividad de una molécula específica de membrana celular que es responsable del movimiento de los precursores de peptidoglicanos desde el citoplasma hacia el exterior de la célula, impidiendo finalmente su incorporación a la pared celular. La bacitracina es efectiva contra una amplia gama de bacterias, incluyendo organismos grampositivos que se encuentran en la piel, como Staphylococcus y Streptococcus. Aunque puede administrarse por vía oral o intramuscular en algunas circunstancias, se ha demostrado que la bacitracina es nefrotóxica (perjudicial para los riñones). Por lo tanto, se combina más comúnmente con neomicina y polimixina en ungüentos tópicos como Neosporina.
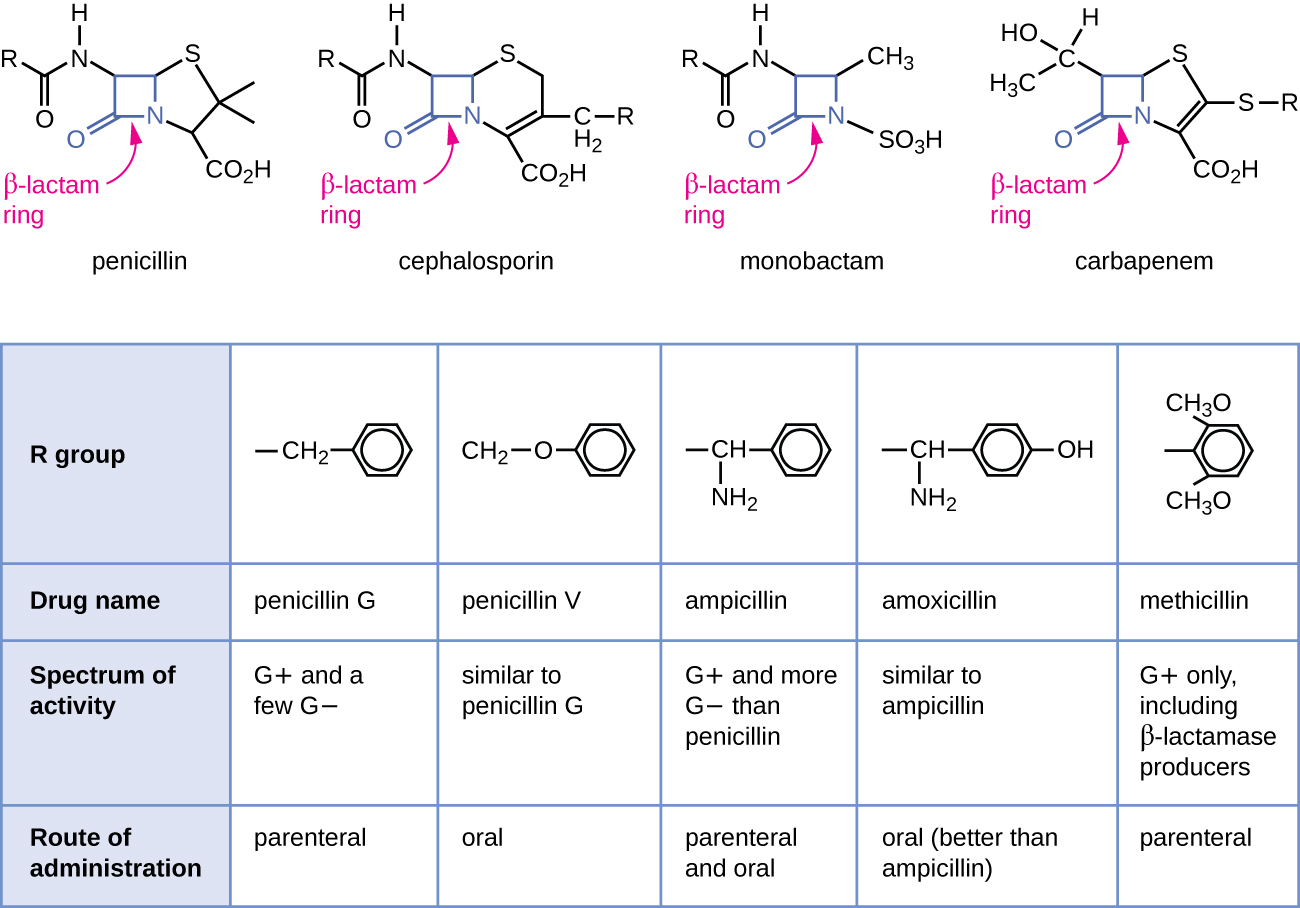
| Mecanismo de Acción | Clase de medicamento | Fármacos Específicos | Natural o Semisintético | Espectro de actividad |
|---|---|---|---|---|
| Interactuar directamente con PBP e inhibir la actividad transpeptidasa | Penicillinas | Penicilina G, penicilina V | Natural | Espectro estrecho contra bacterias grampositivas y algunas bacterias gramnegativas |
| Ampicilina, amoxicilina | Semisintético | Espectro estrecho frente a bacterias grampositivas pero con aumento del espectro gramnegativo | ||
| Meticilina | Semisintético | Espectro estrecho solo contra bacterias grampositivas, incluidas las cepas productoras de penicilinasa | ||
| Cefalosporinas | Cefalosporina C | Natural | Espectro estrecho similar a la penicilina pero con aumento del espectro gramnegativo | |
| Cefalosporinas de primera generación | Semisintético | Espectro estrecho similar a cefalosporina C | ||
| Cefalosporinas de segunda generación | Semisintético | Espectro estrecho pero con espectro gramnegativo aumentado en comparación con la primera generación | ||
| Cefalosporinas de tercera y cuarta generación | Semisintético | Amplio espectro contra bacterias grampositivas y gramnegativas, incluyendo algunos productores de β-lactamasa | ||
| Cefalosporinas de quinta generación | Semisintético | Amplio espectro contra bacterias grampositivas y gramnegativas, incluyendo MRSA | ||
| Monobactámicos | Aztreonam | Semisintético | Espectro estrecho contra bacterias gramnegativas, incluyendo algunos productores de β-lactamasa | |
| Carbapenémicos | Imipenem, meropenem, doripenem | Semisintético | El espectro más amplio de β-lactamas contra bacterias grampositivas y gramnegativas, incluyendo muchos productores de β-lactamasa | |
| Moléculas grandes que se unen a la cadena peptídica de las subunidades de peptidoglicano, bloqueando la transglicosilación y transpeptidación | Glucopéptidos | Vancomicina | Natural | Espectro estrecho solo contra bacterias grampositivas, incluidas las cepas resistentes a múltiples fármacos |
| Bloquear el transporte de subunidades de peptidoglicano a través de la membrana citoplasmática | Bacitracina | Bacitracina | Natural | Amplio espectro contra bacterias grampositivas y gramnegativas |
Ejercicio\(\PageIndex{1}\)
Describir el modo de acción de las β-lactamas.
Inhibidores de la biosíntesis de proteínas
Los ribosomas citoplásmicos que se encuentran en las células animales (80S) son estructuralmente distintos de los que se encuentran en las células bacterianas (70S), haciendo de la biosíntesis de proteínas una buena diana selectiva para los fármacos antibacterianos. Varios tipos de inhibidores de la biosíntesis de proteínas se discuten en esta sección y se resumen en la Figura\(\PageIndex{3}\).
Inhibidores de la síntesis de proteínas que unen la subunidad
Los aminoglucósidos son fármacos antibacterianos grandes y altamente polares que se unen a la subunidad 30S de los ribosomas bacterianos, perjudicando la capacidad correctora del complejo ribosómico. Este deterioro provoca desapareamientos entre codones y anticodones, resultando en la producción de proteínas con aminoácidos incorrectos y proteínas acortadas que se insertan en la membrana citoplásmica. La alteración de la membrana citoplásmica por las proteínas defectuosas mata las células bacterianas. Los aminoglucósidos, que incluyen fármacos como estreptomicina, gentamicina, neomicina y kanamicina, son potentes antibacterianos de amplio espectro. Sin embargo, se ha demostrado que los aminoglucósidos son nefrotóxicos (dañinos para los riñones), neurotóxicos (dañinos para el sistema nervioso) y ototóxicos (dañinos para el oído).
Otra clase de compuestos antibacterianos que se unen a la subunidad 30S son las tetraciclinas. A diferencia de los aminoglucósidos, estos fármacos son bacteriostáticos e inhiben la síntesis de proteínas al bloquear la asociación de los ARNt con el ribosoma durante la traducción. Las tetraciclinas naturales producidas por diversas cepas de Streptomyces se descubrieron por primera vez en la década de 1940, y también se han producido varias tetraciclinas semisintéticas, incluyendo doxiciclina y tigeciclina. Aunque las tetraciclinas son de amplio espectro en su cobertura de patógenos bacterianos, los efectos secundarios que pueden limitar su uso incluyen fototoxicidad, decoloración permanente de dientes en desarrollo y toxicidad hepática con dosis altas o en pacientes con insuficiencia renal.
Inhibidores de síntesis de proteínas que unen la subunidad 50
Existen varias clases de fármacos antibacterianos que funcionan a través de la unión a la subunidad 50S de los ribosomas bacterianos. Los fármacos antibacterianos macrólidos tienen una estructura de anillo grande y compleja y forman parte de una clase más grande de metabolitos secundarios producidos naturalmente llamados policétidos, compuestos complejos producidos de manera escalonada a través de la adición repetida de unidades de dos carbonos por un mecanismo similar al utilizado para la grasa síntesis de ácido. Los macrólidos son fármacos bacteriostáticos de amplio espectro que bloquean la elongación de proteínas al inhibir la formación de enlaces peptídicos entre combinaciones específicas de aminoácidos. El primer macrólido fue eritromicina. Se aisló en 1952 de Streptomyces erythreus y previene la translocación. Los macrólidos semisintéticos incluyen azitromicina y telitromicina. En comparación con la eritromicina, la azitromicina tiene un espectro de actividad más amplio, menos efectos secundarios y una vida media significativamente más larga (1.5 horas para la eritromicina frente a 68 horas para la azitromicina) que permite una dosificación una vez al día y un ciclo corto de terapia de 3 días (es decir, formulación de Zpac) para la mayoría de las infecciones. La telitromicina es el primer semisintético dentro de la clase conocida como cetolidos. Aunque la telitromicina muestra una mayor potencia y actividad contra patógenos resistentes a macrólidos, la Administración de Alimentos y Medicamentos de Estados Unidos (FDA) ha limitado su uso al tratamiento de la neumonía adquirida en la comunidad y requiere la etiqueta más fuerte de “advertencia de caja negra” para el medicamento debido a la grave hepatotoxicidad.
Las lincosamidas incluyen la lincomicina producida naturalmente y la clindamicina semisintética. Aunque estructuralmente distintas de los macrólidos, las lincosamidas son similares en su modo de acción a los macrólidos mediante la unión a la subunidad ribosómica 50S y la prevención de la formación de enlaces peptídicos. Las lincosamidas son particularmente activas contra las infecciones estreptocócicas y estafilocócicas.
El fármaco cloranfenicol representa otra clase estructuralmente distinta de antibacterianos que también se unen al ribosoma 50S, inhibiendo la formación de enlaces peptídicos. El cloranfenicol, producido por Streptomyces venezuelae, fue descubierto en 1947; en 1949, se convirtió en el primer antibiótico de amplio espectro aprobado por la FDA. Aunque es un antibiótico natural, también se sintetiza fácilmente y fue el primer fármaco antibacteriano producido sintéticamente en masa. Como resultado de su producción masiva, cobertura de amplio espectro y capacidad de penetrar en los tejidos de manera eficiente, el cloranfenicol se utilizó históricamente para tratar una amplia gama de infecciones, desde meningitis hasta fiebre tifoidea y conjuntivitis. Desafortunadamente, los efectos secundarios graves, como el síndrome del bebé gris letal y la supresión de la producción de médula ósea, han limitado su papel clínico. El cloranfenicol también causa anemia de dos maneras diferentes. Un mecanismo implica la focalización de ribosomas mitocondriales dentro de las células madre hematopoyéticas, causando una supresión reversible dependiente de la dosis de la producción de células sanguíneas. Una vez que se interrumpe la dosificación de cloranfenicol, la producción de células sanguíneas vuelve a la normalidad. Este mecanismo destaca la similitud entre los ribosomas de bacterias de los 70 y los ribosomas de los 70 dentro de nuestras mitocondrias. El segundo mecanismo de la anemia es idiosincrásico (es decir, el mecanismo no se entiende), e implica una pérdida letal irreversible de la producción de células sanguíneas conocida como anemia aplásica. Este mecanismo de anemia aplásica no depende de la dosis y puede desarrollarse después de que la terapia haya cesado. Debido a preocupaciones de toxicidad, el uso de cloranfenicol en humanos ahora es raro en los Estados Unidos y se limita a infecciones graves que no pueden ser tratadas con antibióticos menos tóxicos. Debido a que sus efectos secundarios son mucho menos graves en animales, se utiliza en medicina veterinaria.
Las oxazolidinonas, incluyendo linezolid, son una nueva clase de inhibidores sintéticos de la síntesis de proteínas de amplio espectro que se unen a la subunidad ribosómica 50S de bacterias grampositivas y gramnegativas. Sin embargo, su mecanismo de acción parece algo diferente al de los otros inhibidores de la síntesis de proteínas de unión a subunidades 50S ya discutidos. En cambio, parecen interferir con la formación del complejo de iniciación (asociación de la subunidad 50S, subunidad 30S y otros factores) para la traducción, y evitan la translocación de la proteína en crecimiento del sitio ribosómico A al sitio P. La tabla\(\PageIndex{3}\) resume los inhibidores de la síntesis de proteínas.
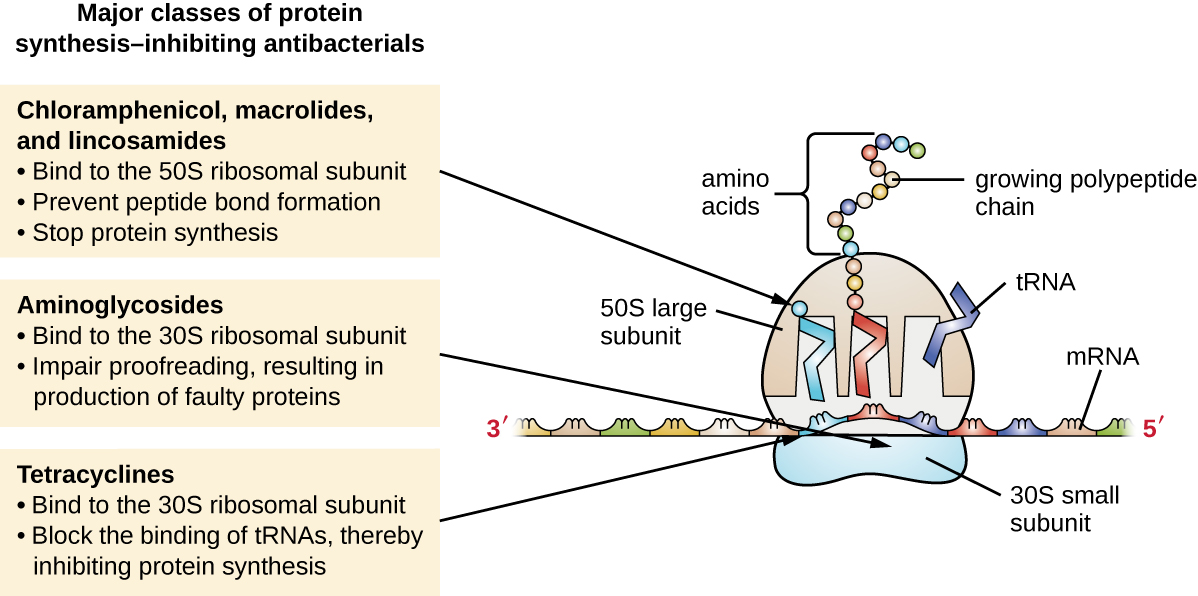
| Objetivo Molecular | Mecanismo de Acción | Clase de medicamento | Fármacos Específicos | Bacteriostático o Bactericida | Espectro de actividad |
|---|---|---|---|---|---|
| Subunidad 30S | Provoca desapareamientos entre codones y anticodones, lo que lleva a proteínas defectuosas que se insertan en la membrana citoplasmática e interrumpen | Aminoglucósidos | Estreptomicina, gentamicina, neomicina, kanamicina | Bactericida | Amplio espectro |
| Bloquea la asociación de ARNt con ribosoma | Tetraciclinas | Tetraciclina, doxiciclina, tigeciclina | Bacteriostático | Amplio espectro | |
| Subunidad 50S | Bloquea la formación de enlaces peptídicos entre aminoácidos | Macrólidos | Eritromicina, azitromicina, telitromicina | Bacteriostático | Amplio espectro |
| Lincosamidas | Lincomicina, clindamicina | Bacteriostático | Espectro estrecho | ||
| No aplica | Cloranfenicol | Bacteriostático | Amplio espectro | ||
| Interfiere con la formación del complejo de iniciación entre las subunidades 50S y 30S y otros factores. | Oxazolidinonas | Linezolid | Bacteriostático | Amplio espectro |
Ejercicio\(\PageIndex{2}\)
Comparar y contrastar los diferentes tipos de inhibidores de la síntesis de proteínas.
Inhibidores de la función membrana
Un pequeño grupo de antibacterianos se dirige a la membrana bacteriana como su modo de acción (Tabla\(\PageIndex{4}\)). Las polimixinas son antibióticos polipeptídicos naturales que se descubrieron por primera vez en 1947 como productos de Bacillus polymyxa; solo se han utilizado clínicamente la polimixina B y la polimixina E (colistina). Son lipofílicas con propiedades similares a detergentes e interactúan con el componente lipopolisacárido de la membrana externa de las bacterias gramnegativas, en última instancia, alterando tanto sus membranas externas como internas y matando las células bacterianas. Desafortunadamente, el mecanismo dirigido a la membrana no es una toxicidad selectiva, y estos fármacos también se dirigen y dañan la membrana de las células en el riñón y el sistema nervioso cuando se administran sistémicamente. Debido a estos graves efectos secundarios y su mala absorción del tracto digestivo, la polimixina B se usa en ungüentos antibióticos tópicos de venta libre (por ejemplo, Neosporina), y la colistina oral se usó históricamente solo para la descontaminación intestinal para prevenir infecciones originadas por microbios intestinales en pacientes inmunodeprimidos o para aquellos sometidos a ciertas cirugías abdominales. Sin embargo, la aparición y propagación de patógenos multirresistentes ha llevado a un mayor uso de colistina intravenosa en los hospitales, a menudo como fármaco de último recurso para tratar infecciones graves. La daptomicina antibacteriana es un lipopéptido cíclico producido por Streptomyces roseosporus que parece funcionar como las polimixinas, insertándose en la membrana celular bacteriana e interrumpiéndola. Sin embargo, en contraste con la polimixina B y la colistina, que se dirigen solo a bacterias gramnegativas, la daptomicina se dirige específicamente a bacterias grampositivas. Por lo general, se administra por vía intravenosa y parece ser bien tolerada, mostrando toxicidad reversible en los músculos esqueléticos.
| Mecanismo de Acción | Clase de medicamento | Fármacos Específicos | Espectro de actividad | Uso Clínico |
|---|---|---|---|---|
| Interactúa con lipopolisacáridos en la membrana externa de bacterias gramnegativas, destruyendo la célula a través de la eventual alteración de la membrana externa y la membrana citoplásmica | Polimixinas | Polimixina B | Espectro estrecho contra bacterias gramnegativas, incluyendo cepas resistentes a múltiples fármacos | Preparaciones tópicas para prevenir infecciones en heridas |
| Polimixina E (colistina) | Espectro estrecho contra bacterias gramnegativas, incluyendo cepas resistentes a múltiples fármacos | Dosificación oral para descontaminar el intestino para prevenir infecciones en pacientes inmunodeprimidos o pacientes sometidos a cirugía/procedimientos invasivos. | ||
| Dosificación intravenosa para tratar infecciones sistémicas graves causadas por patógenos multirresistentes | ||||
| Se inserta en la membrana citoplasmática de bacterias grampositivas, alterando la membrana y matando la célula | Lipopéptido | Daptomicina | Espectro estrecho contra bacterias grampositivas, incluyendo cepas resistentes a múltiples fármacos | Infecciones complicadas de la piel y la estructura de la piel y bacteriemia causadas por patógenos grampositivos, incluyendo MRSA |
Ejercicio\(\PageIndex{3}\)
¿Cómo inhiben las polimixinas la función de la membrana?
Inhibidores de la síntesis de ácidos nucleicos
Algunos fármacos antibacterianos funcionan inhibiendo la síntesis de ácidos nucleicos (Tabla\(\PageIndex{5}\)). Por ejemplo, el metronidazol es un miembro semisintético de la familia de los nitroimidazoles que también es un antiprotozoo. Interfiere con la replicación del ADN en las células diana. El fármaco rifampicina es un miembro semisintético de la familia de la rifamicina y funciona bloqueando la actividad de la ARN polimerasa en bacterias. Las enzimas ARN polimerasa en bacterias son estructuralmente diferentes de las de los eucariotas, proporcionando toxicidad selectiva contra células bacterianas. Se utiliza para el tratamiento de una variedad de infecciones, pero su uso primario, a menudo en un cóctel con otros fármacos antibacterianos, es contra las micobacterias que causan tuberculosis. A pesar de la selectividad de su mecanismo, la rifampicina puede inducir a las enzimas hepáticas a incrementar el metabolismo de otros fármacos que se administran (antagonismo), dando lugar a hepatotoxicidad (toxicidad hepática) e influyendo negativamente en la biodisponibilidad y efecto terapéutico de los fármacos acompañantes.
Un miembro de la familia de las quinolonas, un grupo de antimicrobianos sintéticos, es el ácido nalidíxico. Fue descubierto en 1962 como subproducto durante la síntesis de cloroquina, un fármaco antipalúdico. El ácido nalidíxico inhibe selectivamente la actividad de la ADN girasa bacteriana, bloqueando la replicación del ADN. Las modificaciones químicas en el esqueleto original de quinolonas han dado como resultado la producción de fluoroquinolonas, como ciprofloxacino y levofloxacino, que también inhiben la actividad de la ADN girasa. Ciprofloxacino y levofloxacino son eficaces contra un amplio espectro de bacterias grampositivas o gramnegativas, y se encuentran entre los antibióticos más comúnmente recetados utilizados para tratar una amplia gama de infecciones, incluyendo infecciones del tracto urinario, infecciones respiratorias, infecciones abdominales e infecciones de la piel. Sin embargo, a pesar de su toxicidad selectiva contra la ADN girasa, los efectos secundarios asociados a diferentes fluoroquinolonas incluyen fototoxicidad, neurotoxicidad, cardiotoxicidad, disfunción del metabolismo de la glucosa y aumento del riesgo de ruptura del tendón.
| Mecanismos de Acción | Clase de medicamento | Fármacos Específicos | Espectro de actividad | Uso Clínico |
|---|---|---|---|---|
| Inhibe la actividad de la ARN polimerasa bacteriana y bloquea la transcripción, matando la célula | Rifamicina | Rifampina | Espectro estrecho con actividad frente a bacterias grampositivas y números limitados de bacterias gramnegativas. También activo contra Mycobacterium tuberculosis. | Terapia combinada para el tratamiento de la tuberculosis |
| Inhibe la actividad de la ADN girasa y bloquea la replicación del ADN, matando la célula | Fluoroquinolonas | Ciprofloxacino, ofloxacino, moxifloxacino | Amplio espectro contra bacterias grampositivas y gramnegativas | Amplia variedad de infecciones cutáneas y sistémicas |
Ejercicio\(\PageIndex{4}\)
¿Por qué los inhibidores de la síntesis bacteriana de ácidos nucleicos no se dirigen a las células huésped
Inhibidores de vías metabólicas
Algunos fármacos sintéticos controlan las infecciones bacterianas al funcionar como antimetabolitos, inhibidores competitivos para las enzimas metabólicas bacterianas (Tabla\(\PageIndex{6}\)). Las sulfonamidas (sulfamidas) son los agentes antibacterianos sintéticos más antiguos y son análogos estructurales del ácido para-aminbenzoico (PABA), un intermedio temprano en la síntesis de ácido fólico (Figura\(\PageIndex{4}\)). Al inhibir la enzima involucrada en la producción de ácido dihidrofólico, las sulfonamidas bloquean la biosíntesis bacteriana de ácido fólico y, posteriormente, pirimidinas y purinas requeridas para la síntesis de ácidos nucleicos. Este mecanismo de acción proporciona inhibición bacteriostática del crecimiento contra un amplio espectro de patógenos grampositivos y gramnegativos. Debido a que los humanos obtienen ácido fólico de los alimentos en lugar de sintetizarlo intracelularmente, las sulfonamidas son selectivamente tóxicas para las bacterias. Sin embargo, las reacciones alérgicas a los fármacos sulfa son comunes. Las sulfonas son estructuralmente similares a las sulfonamidas pero no se usan comúnmente hoy en día excepto para el tratamiento de la enfermedad de Hansen (lepra).
La trimetoprima es un compuesto antimicrobiano sintético que sirve como antimetabolito dentro de la misma vía de síntesis de ácido fólico que las sulfonamidas. Sin embargo, la trimetoprima es un análogo estructural del ácido dihidrofólico e inhibe un paso posterior en la vía metabólica (Figura\(\PageIndex{4}\)). El trimetoprima se usa en combinación con el medicamento sulfa sulfametoxazol para tratar infecciones del tracto urinario, infecciones del oído y bronquitis. Como se discutió, la combinación de trimetoprima y sulfametoxazol es un ejemplo de sinergia antibacteriana. Cuando se usa solo, cada antimetabolito solo disminuye la producción de ácido fólico a un nivel donde se produce la inhibición bacteriostática del crecimiento. Sin embargo, cuando se usa en combinación, la inhibición de ambos pasos en la vía metabólica disminuye la síntesis de ácido fólico a un nivel que es letal para la célula bacteriana. Debido a la importancia del ácido fólico durante el desarrollo fetal, los sulfa y el uso de trimetoprima deben considerarse cuidadosamente durante el embarazo temprano.
El fármaco isoniazida es un antimetabolito con toxicidad específica para micobacterias y se ha utilizado durante mucho tiempo en combinación con rifampicina o estreptomicina en el tratamiento de la tuberculosis. Se administra como profármaco, requiriendo activación a través de la acción de una enzima peroxidasa bacteriana intracelular, formando isoniacida-nicotinamida adenina dinucleótido (NAD) e isoniacida-nicotinamida adenina dinucleótido fosfato (NADP), impidiendo en última instancia la síntesis de ácido micólico, que es esencial para las paredes celulares micobacterianas. Los posibles efectos secundarios del uso de isoniazida incluyen hepatotoxicidad, neurotoxicidad y toxicidad hematológica (anemia).
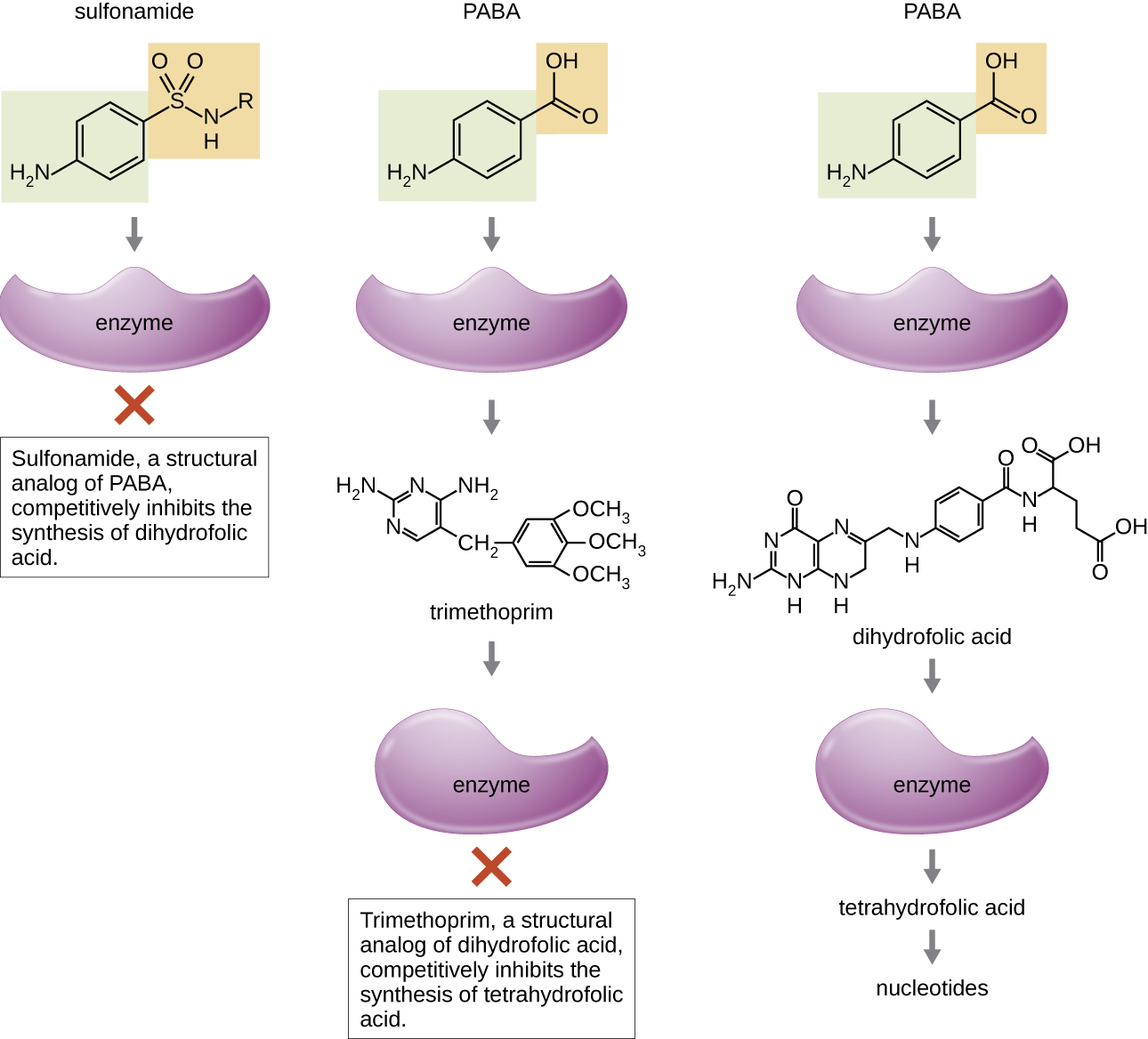
| Objetivo de la vía metabólica | Mecanismo de Acción | Clase de medicamento | Fármacos Específicos | Espectro de actividad |
|---|---|---|---|---|
| Síntesis de ácido fólico | Inhibe la enzima involucrada en la producción de ácido dihidrofólico | Sulfonamidas | Sulfametoxazol | Amplio espectro contra bacterias grampositivas y gramnegativas |
| Sulfonas | Dapsone | |||
| Inhibe la enzima involucrada en la producción de ácido tetrahidrofólico | No aplica | Trimetoprima | Amplio espectro contra bacterias grampositivas y gramnegativas | |
| Síntesis de ácido micólico | Interfiere con la síntesis de ácido micólico | No aplica | Isoniazida | Espectro estrecho contra Mycobacterium spp., incluyendo M. tuberculosis |
Ejercicio\(\PageIndex{5}\)
¿Cómo las sulfonamidas y la trimetoprima se dirigen selectivamente a las bacterias?
Inhibidor de la ATP Sintasa
La bedaquilina, que representa la clase de compuestos antibacterianos sintéticos llamados diarilquinolonas, utiliza un nuevo modo de acción que inhibe específicamente el crecimiento micobacteriano. Aunque aún no se ha aclarado el mecanismo específico, este compuesto parece interferir con la función de las ATP sintasas, tal vez interfiriendo con el uso del gradiente de iones hidrógeno para la síntesis de ATP por fosforilación oxidativa, conduciendo a una producción reducida de ATP. Debido a sus efectos secundarios, incluyendo hepatotoxicidad y arritmia cardíaca potencialmente letal, su uso está reservado para casos graves, por lo demás intratables de tuberculosis.
Enfoque Clínico: Parte 2
Al leer minuciosamente los antecedentes de salud de Marisa, la doctora notó que durante su hospitalización en Vietnam, fue cateterizada y recibió los medicamentos antimicrobianos ceftazidima y metronidazol. Al enterarse de esto, el médico ordenó una tomografía computarizada del abdomen de Marisa para descartar apendicitis; el médico también solicitó análisis de sangre para ver si tenía un recuento elevado de glóbulos blancos, y ordenó un análisis de orina y un cultivo de orina para buscar la presencia de glóbulos blancos, glóbulos rojos y bacterias .
La muestra de orina de Marisa dio positivo por la presencia de bacterias, lo que indica una infección del tracto urinario (ITU). El médico le recetó ciprofloxacino. Mientras tanto, su orina fue cultivada para cultivar la bacteria para realizar más pruebas.
Ejercicio\(\PageIndex{6}\)
- ¿Qué tipos de antimicrobianos se recetan típicamente para las infecciones urinarias?
- Con base en los antimicrobianos que le dieron en Vietnam, ¿cuál de los antimicrobianos para el tratamiento de una ITU pronosticaría que sería ineficaz?
Conceptos clave y resumen
- Los compuestos antibacterianos presentan toxicidad selectiva, en gran parte debido a las diferencias entre la estructura celular procariota y eucariota.
- Los inhibidores de la síntesis de la pared celular, incluyendo las β-lactamas, los glicopéptidos y la bacitracina, interfieren con la síntesis de peptidoglicanos, haciendo que las células bacterianas sean más propensas a la lisis osmótica
- Hay una variedad de inhibidores de la síntesis de proteínas bacterianas de amplio espectro que se dirigen selectivamente al ribosoma procariota 70S, incluyendo aquellos que se unen a la subunidad 30S (aminoglucósidos y tetraciclinas) y otros que se unen a la subunidad 50S (macrólidos, lincosamidas, cloranfenicol y oxazolidinonas).
- Las polimixinas son antibióticos polipeptídicos lipófilos que se dirigen al componente lipopolisacárido de bacterias gramnegativas y finalmente alteran la integridad de las membranas externas e internas de estas bacterias.
- Los inhibidores de la síntesis de ácidos nucleicos rifamicinas y fluoroquinolonas se dirigen a la transcripción del ARN bacteriano y la replicación del ADN, respectivamente.
- Algunos fármacos antibacterianos son antimetabolitos, actuando como inhibidores competitivos de las enzimas metabólicas bacterianas. Las sulfonamidas y la trimetoprima son antimetabolitos que interfieren con la síntesis bacteriana de ácido fólico. La isoniazida es un antimetabolito que interfiere con la síntesis de ácido micólico en micobacterias.