
14.4: Consideraciones clínicas
- Page ID
- 54917

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Objetivos de aprendizaje
- Explicar las diferencias entre los modos de acción de los fármacos que se dirigen a hongos, protozoos, helmintos y virus
Debido a que los hongos, protozoos y helmintos son eucariotas, sus células son muy similares a las células humanas, lo que dificulta el desarrollo de fármacos con toxicidad selectiva. Adicionalmente, los virus se replican dentro de las células hospedadoras humanas, lo que dificulta el desarrollo de fármacos que son selectivamente tóxicos para virus o células infectadas por virus. A pesar de estos desafíos, existen medicamentos antimicrobianos que se dirigen a hongos, protozoos, helmintos y virus, y algunos incluso se dirigen a más de un tipo de microbio. La Tabla\(\PageIndex{1}\), la Tabla\(\PageIndex{2}\)\(\PageIndex{3}\), la Tabla y la Tabla\(\PageIndex{4}\) proporcionan ejemplos de medicamentos antimicrobianos en estas diversas clases.
Fármacos Antifúngicos
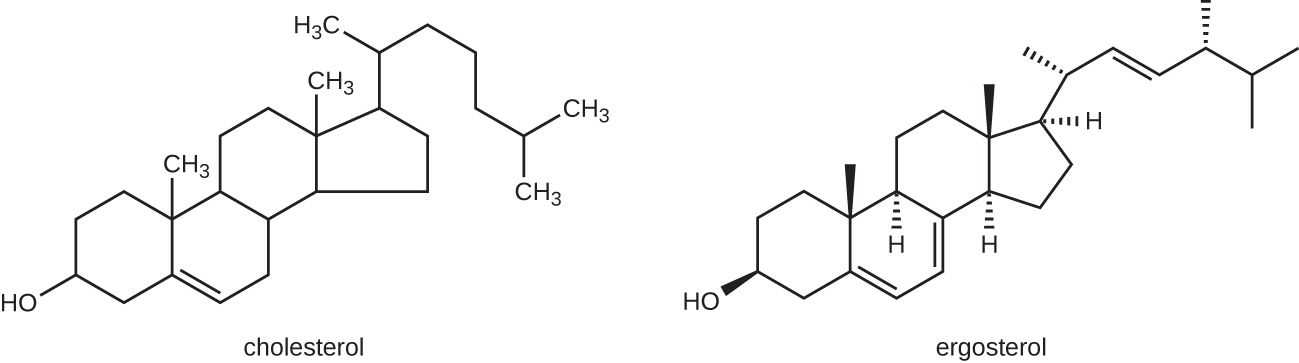
El modo de acción más común para los fármacos antifúngicos es la alteración de la membrana celular. Los antifúngicos aprovechan pequeñas diferencias entre hongos y humanos en las vías bioquímicas que sintetizan esteroles. Los esteroles son importantes para mantener la fluidez adecuada de la membrana y, por lo tanto, el correcto funcionamiento de la membrana celular. Para la mayoría de los hongos, el esterol de membrana predominante es el ergosterol. Debido a que las membranas celulares humanas usan colesterol, en lugar de ergosterol, los fármacos antifúngicos que se dirigen a la síntesis de ergosterol son selectivamente tóxicos (Figura\(\PageIndex{1}\)).
Los imidazoles son fungicidas sintéticos que interrumpen la biosíntesis del ergosterol; se utilizan comúnmente en aplicaciones médicas y también en agricultura para evitar que las semillas y los cultivos cosechados se moldeen. Los ejemplos incluyen miconazol, ketoconazol y clotrimazol, que se usan para tratar infecciones fúngicas de la piel como la tiña, específicamente la tiña pedis (pie de atleta), la tiña cruris (tiña inguinal) y la tiña corporal. Estas infecciones son comúnmente causadas por dermatofitos de los géneros Trichophyton, Epidermophyton y Microsporum. El miconazol también se usa predominantemente para el tratamiento de las infecciones vaginales causadas por el hongo Candida, y el ketoconazol se usa para el tratamiento de la tiña versicolor y la caspa, que ambos pueden ser causados por el hongo Malassezia.
Los fármacos de triazol, incluido el fluconazol, también inhiben la biosíntesis del ergosterol. Sin embargo, se pueden administrar por vía oral o intravenosa para el tratamiento de varios tipos de infecciones sistémicas por levaduras, entre ellas la candidiasis oral y la meningitis criptocócica, ambas prevalentes en pacientes con SIDA. Los triazoles también presentan una toxicidad más selectiva, en comparación con los imidazoles, y se asocian con menos efectos secundarios.
Las alilaminas, una clase estructuralmente diferente de fármacos antifúngicos sintéticos, inhiben un paso más temprano en la biosíntesis del ergosterol. La alilamina más utilizada es la terbinafina (comercializada bajo la marca Lamisil), que se usa tópicamente para el tratamiento de infecciones dermatofíticas de la piel como pie de atleta, tiña y tiña inguinal. El tratamiento oral con terbinafina también se usa para el tratamiento de hongos en las uñas de las manos y los pies, pero puede asociarse con el raro efecto secundario de la hepatotoxicidad.
Los polienos son una clase de agentes antifúngicos producidos naturalmente por ciertas bacterias actinomicetos del suelo y están estructuralmente relacionados con macrólidos. Estas grandes moléculas lipofílicas se unen al ergosterol en las membranas citoplásmicas fúngicas, creando así poros. Los ejemplos comunes incluyen nistatina y anfotericina B. La nistatina se usa típicamente como tratamiento tópico para infecciones por levaduras de la piel, la boca y la vagina, pero también se puede usar para infecciones fúngicas intestinales. El medicamento anfotericina B se usa para infecciones fúngicas sistémicas como aspergilosis, meningitis criptocócica, histoplasmosis, blastomicosis y candidiasis. La anfotericina B fue el único fármaco antifúngico disponible durante varias décadas, pero su uso se asocia a algunos efectos secundarios graves, entre ellos la nefrotoxicidad (toxicidad renal).
La anfotericina B se usa a menudo en combinación con flucitosina, un análogo de pirimidina fluorada que es convertido por una enzima específica de hongos en un producto tóxico que interfiere tanto con la replicación del ADN como en la síntesis de proteínas en hongos. La flucitosina también se asocia con hepatotoxicidad (toxicidad hepática) y depresión de la médula ósea.
Más allá de dirigirse al ergosterol en las membranas celulares fúngicas, hay algunos fármacos antifúngicos que se dirigen a otras estructuras fúngicas (Figura\(\PageIndex{2}\)). Las equinocandinas, incluida la caspofungina, son un grupo de compuestos antifúngicos producidos naturalmente que bloquean la síntesis de β (1→3) glucano que se encuentra en las paredes celulares fúngicas pero que no se encuentra en las células humanas. Esta clase de drogas tiene el apodo de “penicilina para hongos”. La caspofungina se utiliza para el tratamiento de la aspergilosis así como de las infecciones sistémicas por levaduras.
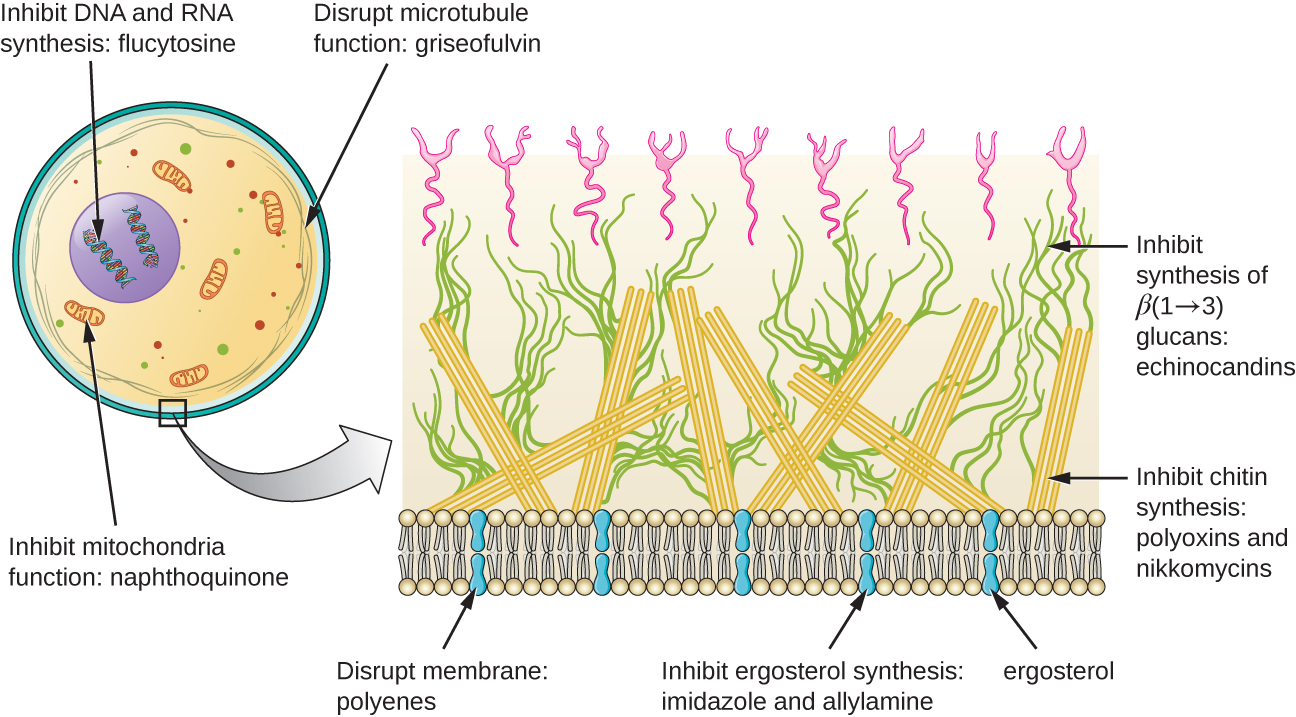
Aunque la quitina es solo un constituyente menor de las paredes celulares fúngicas, también está ausente en las células humanas, convirtiéndola en una diana selectiva. Las polioxinas y nikkomicinas son antifúngicos producidos naturalmente que se dirigen a la síntesis de quitina. Las polioxinas se utilizan para controlar hongos con fines agrícolas, y la nikkomicina Z está actualmente en desarrollo para su uso en humanos para tratar infecciones por levaduras y fiebre del Valle (coccidioidomicosis), una enfermedad fúngica prevalente en el suroeste de Estados Unidos. 1
Se cree que la griseofulvina antifúngica producida naturalmente altera específicamente la división celular fúngica al interferir con los microtúbulos involucrados en la formación del huso durante la mitosis. Fue uno de los primeros antifúngicos, pero su uso se asocia a hepatotoxicidad. Por lo general, se administra por vía oral para tratar diversos tipos de infecciones dermatofíticas de la piel cuando otros tratamientos antifúngicos tópicos son ineficaces.
Existen algunos fármacos que actúan como antimetabolitos contra los procesos fúngicos. Por ejemplo, la atovaquona, un representante de la clase de fármacos de la naftoquinona, es un antimetabolito semisintético para versiones fúngicas y protozoarias de un citocromo mitocondrial importante en el transporte de electrones. Estructuralmente, es un análogo de la coenzima Q, con la que compite por la unión electrónica. Es particularmente útil para el tratamiento de la neumonía por Pneumocystis causada por Pneumocystis jirovecii. La combinación antibacteriana sulfametoxazol-trimetoprima también actúa como antimetabolito contra P. jirovecii.
En el cuadro se\(\PageIndex{1}\) muestran las diversas clases terapéuticas de fármacos antifúngicos, categorizados por modo de acción, con ejemplos de cada uno.
| Mecanismo de Acción | Clase de medicamento | Fármacos Específicos | Usos Clínicos |
|---|---|---|---|
| Inhibe la síntesis de ergosterol | Imidazoles | Miconazol, ketoconazol, clotrimazol | Infecciones fúngicas de la piel e infecciones vaginales |
| Triazoles | Fluconazol | Infecciones sistémicas por levaduras, aftas orales y meningitis criptocócica | |
| Alilaminas | Terbinafina | Infecciones dermatofíticas de la piel (pie de atleta, gusano anular, tiña inguinal) e infecciones de las uñas de las manos y los pies | |
| Unir ergosterol en la membrana celular y crear poros que interrumpen la membrana | Polienos | Nistatina | Se usa tópicamente para infecciones por hongos en la piel, la boca y la vagina; también se usa para infecciones fúngicas del intestino |
| Anfotericina B | Variedad de infecciones fúngicas sistémicas | ||
| Inhibe la síntesis de paredes | Echinocandinas | Caspofungina | Aspergilosis e infecciones sistémicas por levaduras |
| No aplica | Nikkomicina Z | Coccidioidomicosis (fiebre del valle) e infecciones por levaduras | |
| Inhibe microtúbulos y división celular | No aplica | Griseofulvina | Infecciones dermatofíticas de la piel |
Ejercicio\(\PageIndex{1}\)
¿Cómo es la interrupción de la biosíntesis de ergosterol un modo efectivo de acción para los antifúngicos?
Tratamiento de una infección fúngica de los pulmones
Jack, un ingeniero de 48 años, es VIH positivo pero generalmente saludable gracias a la terapia antirretroviral (TAR). No obstante, después de una semana particularmente intensa en el trabajo, desarrolló fiebre y tos seca. Asumió que solo tenía una gripe fría o leve por sobreesfuerzo y no pensó mucho en ello. Sin embargo, después de aproximadamente una semana, comenzó a experimentar fatiga, pérdida de peso y dificultad para respirar. Decidió visitar a su médico, quien encontró que Jack tenía un bajo nivel de oxigenación sanguínea. El médico ordenó análisis de sangre, una radiografía de tórax y la recolección de una muestra de esputo inducido para su análisis. Su radiografía mostró una fina turbidez y varios neumatoceles (bolsas de aire de pared delgada), lo que indicó neumonía por Pneumocystis (PCP), un tipo de neumonía causada por el hongo Pneumocystis jirovecii. El médico de Jack lo ingresó en el hospital y le recetó Bactrim, una combinación de sulfametoxazol y trimetoprima, para ser administrado por vía intravenosa.
P. jirovecii es un hongo similar a levadura con un ciclo de vida similar al de los protozoos. Como tal, se clasificó como protozoo hasta la década de 1980. Vive sólo en el tejido pulmonar de las personas infectadas y se transmite de persona a persona, con muchas personas expuestas cuando eran niños. Típicamente, P. jirovecii solo causa neumonía en individuos inmunodeprimidos. Las personas sanas pueden portar el hongo en sus pulmones sin síntomas de enfermedad. La PCP es particularmente problemática entre los pacientes con VIH con sistemas inmunitarios comprometidos.
El PCP generalmente se trata con Bactrim oral o intravenoso, pero la atovacuona o pentamidina (otro fármaco antiparasitario) son alternativas. Si no se trata, el PCP puede progresar, lo que lleva a un colapso pulmonar y una mortalidad de casi el 100%. Incluso con la terapia con medicamentos antimicrobianos, el PCP sigue siendo responsable del 10% de las muertes relacionadas con el VIH.
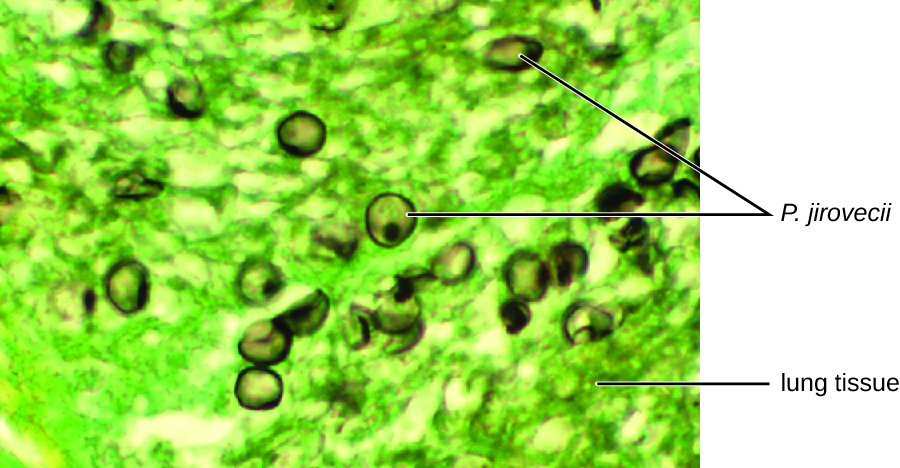
El examen citológico, mediante el ensayo de inmunofluorescencia directa (DFA), de un frotis de una muestra de esputo de Jack confirmó la presencia de P. jirovecii (Figura\(\PageIndex{3}\)). Adicionalmente, los resultados de los análisis de sangre de Jack revelaron que su recuento de glóbulos blancos había disminuido, haciéndolo más susceptible al hongo. Su médico revisó su régimen de TAR e hizo ajustes. Después de unos días de hospitalización, Jack fue dado de alta para continuar con su terapia antimicrobiana en casa. Con los ajustes a su terapia ART, los recuentos de CD4 de Jack comenzaron a aumentar y pudo volver a trabajar.
Medicamentos antiprotozoarios
Existen algunos mecanismos por los cuales los fármacos antiprotozoarios se dirigen a los protozoos infecciosos (Tabla\(\PageIndex{3}\)). Algunos son antimetabolitos, como atovaquona, proguanil y artemisininas. La atovacuona, además de ser antifúngica, bloquea el transporte de electrones en protozoos y se utiliza para el tratamiento de infecciones protozoarias incluyendo malaria, babesiosis y toxoplasmosis. El proguanil es otro antimetabolito sintético que se procesa en las células parasitarias en su forma activa, lo que inhibe la síntesis protozoaria de ácido fólico. A menudo se usa en combinación con atovaquona, y la combinación se comercializa como Malarone tanto para el tratamiento como para la prevención de la malaria.
La artemisinina, un antifúngico derivado de plantas descubierto por primera vez por científicos chinos en la década de 1970, es bastante eficaz contra la malaria. Los derivados semisintéticos de la artemisinina son más solubles en agua que la versión natural, lo que los hace más biodisponibles. Aunque el mecanismo exacto de acción no está claro, las artemisininas parecen actuar como profármacos que son metabolizados por las células diana para producir especies reactivas de oxígeno (ROS) que dañan las células diana. Debido al aumento de la resistencia a los medicamentos antipalúdicos, las artemisininas también se usan comúnmente en combinación con otros compuestos antipalúdicos en la terapia de combinación a base de artemisinina (ACT).
Se utilizan varios antimetabolitos para el tratamiento de la toxoplasmosis causada por el parásito Toxoplasma gondii. El fármaco sulfa sintético sulfadiazina inhibe competitivamente una enzima en la producción de ácido fólico en parásitos y puede usarse para tratar la malaria y la toxoplasmosis. La pirimetamina es un fármaco sintético que inhibe una enzima diferente en la vía de producción de ácido fólico y a menudo se usa en combinación con sulfadoxina (otro fármaco de sulfa) para el tratamiento de la malaria o en combinación con sulfadiazina para el tratamiento de la toxoplasmosis. Los efectos secundarios de la pirimetamina incluyen disminución de la actividad de la médula ósea que puede causar aumento de hematomas y recuento bajo de glóbulos rojos. Cuando la toxicidad es una preocupación, la espiramicina, un inhibidor de la síntesis de proteínas macrólidas, se administra típicamente para el tratamiento de la toxoplasmosis.
Dos clases de fármacos antiprotozoarios interfieren con la síntesis de ácidos nucleicos: nitroimidazoles y quinolinas. Los nitroimidazoles, incluyendo metronidazol semisintético, que se discutió previamente como fármaco antibacteriano, y tinidazol sintético, son útiles para combatir una amplia variedad de patógenos protozoarios, como Giardia lamblia, Entamoeba histolytica y Trichomonas vaginalis . Tras la introducción en estas células en ambientes bajos en oxígeno, los nitroimidazoles se activan e introducen la rotura de la cadena del ADN, interfiriendo con la replicación del ADN en las células diana. Desafortunadamente, el metronidazol está asociado con la carcinogénesis (el desarrollo del cáncer) en humanos.
Otro tipo de fármaco antiprotozoario sintético que durante mucho tiempo se ha pensado que interfiere específicamente con la replicación del ADN en ciertos patógenos es la pentamidina. Históricamente se ha utilizado para el tratamiento de la enfermedad del sueño africana (causada por el protozoo Trypanosoma brucei) y la leishmaniasis (causada por protozoos del género Leishmania), pero también es un tratamiento alternativo para el hongo Pneumocystis. Algunos estudios indican que se une específicamente al ADN que se encuentra dentro de los cinetoplastos (ADNk; estructuras largas similares a mitocondrias únicas de los tripanosomas), lo que lleva a la escisión del ADNk. Sin embargo, el ADN nuclear tanto del parásito como del huésped no se ve afectado. También parece unirse al ARNt, inhibiendo la adición de aminoácidos al ARNt, previniendo así la síntesis de proteínas. Los posibles efectos secundarios del uso de pentamidina incluyen disfunción pancreática y daño hepático.
Las quinolinas son una clase de compuestos sintéticos relacionados con la quinina, que tiene una larga historia de uso contra la malaria. Se cree que las quinolinas interfieren con la desintoxicación del hemo, que es necesaria para que el parásito descomponga eficazmente la hemoglobina en aminoácidos dentro de los glóbulos rojos. Los derivados sintéticos cloroquina, quinacrina (también llamada mepacrina) y mefloquina se usan comúnmente como antimaláricos, y la cloroquina también se usa para tratar la amebiasis típicamente causada por Entamoeba histolytica. El uso profiláctico a largo plazo de cloroquina o mefloquina puede provocar efectos secundarios graves, incluyendo alucinaciones o problemas cardíacos. Los pacientes con deficiencia de glucosa-6-fosfato deshidrogenasa experimentan anemia severa cuando se tratan con cloroquina.
| Mecanismo de Acción | Clase de medicamento | Fármacos Específicos | Usos Clínicos |
|---|---|---|---|
| Inhiben el transporte de electrones en mitocondrias | Naftoquinona | Atovacone | Malaria, babesiosis y toxoplasmosis |
| Inhibe la síntesis de ácido | No aplica | Proquanil | Terapia combinada con atovacuona para el tratamiento y prevención de la malaria |
| Sulfonamida | Sulfadiazina | Malaria y toxoplasmosis | |
| No aplica | Pirimetamina | Terapia de combinación con sulfadoxina (fármaco sulfa) para la malaria | |
| Produce especies reactivas dañinas de oxígeno | No aplica | Artemisinina | Terapia combinada para tratar la malaria |
| Inhibe síntesis de ADN | Nitroimidazoles | Metronidazol, tinidazol | Infecciones por Giardia lamblia, Entamoeba histolytica y Trichomonas vaginalis |
| No aplica | Pentamidina | Enfermedad africana del sueño y leishmaniasis | |
| Inhibe la desintoxicación del hemo | Quinolinas | Cloroquina | Malaria e infecciones con E. histolytica |
| Mepacrina, mefloquina | Malaria |
Ejercicio\(\PageIndex{2}\)
Enumerar dos modos de acción para los fármacos antiprotozoarios.
Medicamentos antihelmínticos
Debido a que los helmintos son eucariotas multicelulares como los humanos, desarrollar fármacos con toxicidad selectiva contra ellos es extremadamente desafiante. A pesar de esto, se han desarrollado varias clases efectivas (Tabla\(\PageIndex{3}\)). Los bencimidazoles sintéticos, como el mebendazol y el albendazol, se unen a la β-tubulina helmíntica, previniendo la formación de microtúbulos. Los microtúbulos en las células intestinales de los gusanos parecen estar particularmente afectados, lo que lleva a una reducción en la captación de glucosa. Además de su actividad contra una amplia gama de helmintos, los bencimidazoles también son activos contra muchos protozoos, hongos y virus, y su uso para inhibir la mitosis y la progresión del ciclo celular en células cancerosas está en estudio. 2 Los posibles efectos secundarios de su uso incluyen daño hepático y supresión de la médula ósea.
Las avermectinas son miembros de la familia de los macrólidos que se descubrieron por primera vez a partir de un aislado de suelo japonés, Streptomyces avermectinius. Un derivado semisintético más potente de la avermectina es la ivermectina, que se une a canales de cloruro regulados por glutamato específicos de invertebrados incluyendo helmintos, bloqueando la transmisión neuronal y causando inanición, parálisis y muerte de los gusanos. La ivermectina se usa para tratar enfermedades de lombrices intestinales, incluyendo oncocercosis (también llamada ceguera de río, causada por el gusano Onchocerca volvulus) y estrongiloidiasis (causada por el gusano Strongyloides stercoralis o S. fuelleborni). La ivermectina también puede tratar insectos parásitos como ácaros, piojos y chinches, y no es tóxica para los humanos.
La niclosamida es una droga sintética que se ha utilizado por más de 50 años para tratar infecciones por tenia. Aunque su modo de acción no está del todo claro, la niclosamida parece inhibir la formación de ATP en condiciones anaerobias e inhibir la fosforilación oxidativa en las mitocondrias de sus patógenos diana. La niclosamida no se absorbe del tracto gastrointestinal, por lo que puede lograr altas concentraciones intestinales localizadas en los pacientes. Recientemente, se ha demostrado que también tiene actividades antibacterianas, antivirales y antitumorales. 3 4 5
Otro fármaco antihelmíntico sintético es el praziquantel, que se utiliza para el tratamiento de tenias parasitarias y trematodos hepáticos, y es particularmente útil para el tratamiento de la esquistosomiasis (causada por trematodos sanguíneos de tres géneros de Schistosoma). Su modo de acción sigue sin estar claro, pero parece provocar la afluencia de calcio al gusano, resultando en un intenso espasmo y parálisis del gusano. A menudo se usa como alternativa preferida a la niclosamida en el tratamiento de tenias cuando el malestar gastrointestinal limita el uso de niclosamida.
Las tioxantenonas, otra clase de drogas sintéticas estructuralmente relacionadas con la quinina, exhiben actividad antiesquistosómica inhibiendo la síntesis de ARN. La tioxantenona lucanthona y su metabolito hycanthone fueron los primeros utilizados clínicamente, pero los efectos secundarios neurológicos, gastrointestinales, cardiovasculares y hepáticos graves condujeron a su interrupción. La oxamniquina, un derivado menos tóxico de la himantona, solo es efectiva contra S. mansoni, una de las tres especies conocidas por causar esquistosomiasis en humanos. El praziquantel se desarrolló para apuntar a las otras dos especies de esquistosomas, pero las preocupaciones sobre el aumento de la resistencia han renovado el interés en desarrollar derivados adicionales de oxamniquina para apuntar a las tres especies de esquistosomas clínicamente importantes.
| Mecanismo de Acción | Clase de medicamento | Fármacos Específicos | Usos Clínicos |
|---|---|---|---|
| Inhibe la formación de microtúbulos, reduciendo la captación de | Benzimidazoles | Mebendazol, albendazol | Variedad de infecciones por helmintos |
| Bloquear la transmisión neuronal, causando parálisis e inanición | Avermectinas | Ivermectina | Enfermedades por lombrices intestinales, incluida la ceguera de los ríos y la estrongiloidiasis, y tratamiento de insectos parásitos |
| Inhibe producción de ATP | No aplica | Niclosamida | Infecciones intestinales por tenia |
| Inducir la afluencia de calcio | No aplica | Praziquantel | Esquistosomiasis (duelos sanguíneos) |
| Inhibe la síntesis de | Tioxantenonas | Lucanthone, hycanthone, oxamniquina | Esquistosomiasis (duelos sanguíneos) |
Ejercicio\(\PageIndex{3}\)
¿Por qué los medicamentos antihelmínticos son difíciles de desarrollar?
Medicamentos Antivirales
A diferencia de la compleja estructura de hongos, protozoos y helmintos, la estructura viral es simple, consistente en ácido nucleico, una cubierta proteica, enzimas virales y, a veces, una envoltura lipídica. Además, los virus son patógenos intracelulares obligados que utilizan la maquinaria celular del huésped para replicarse. Estas características dificultan el desarrollo de fármacos con toxicidad selectiva contra virus.
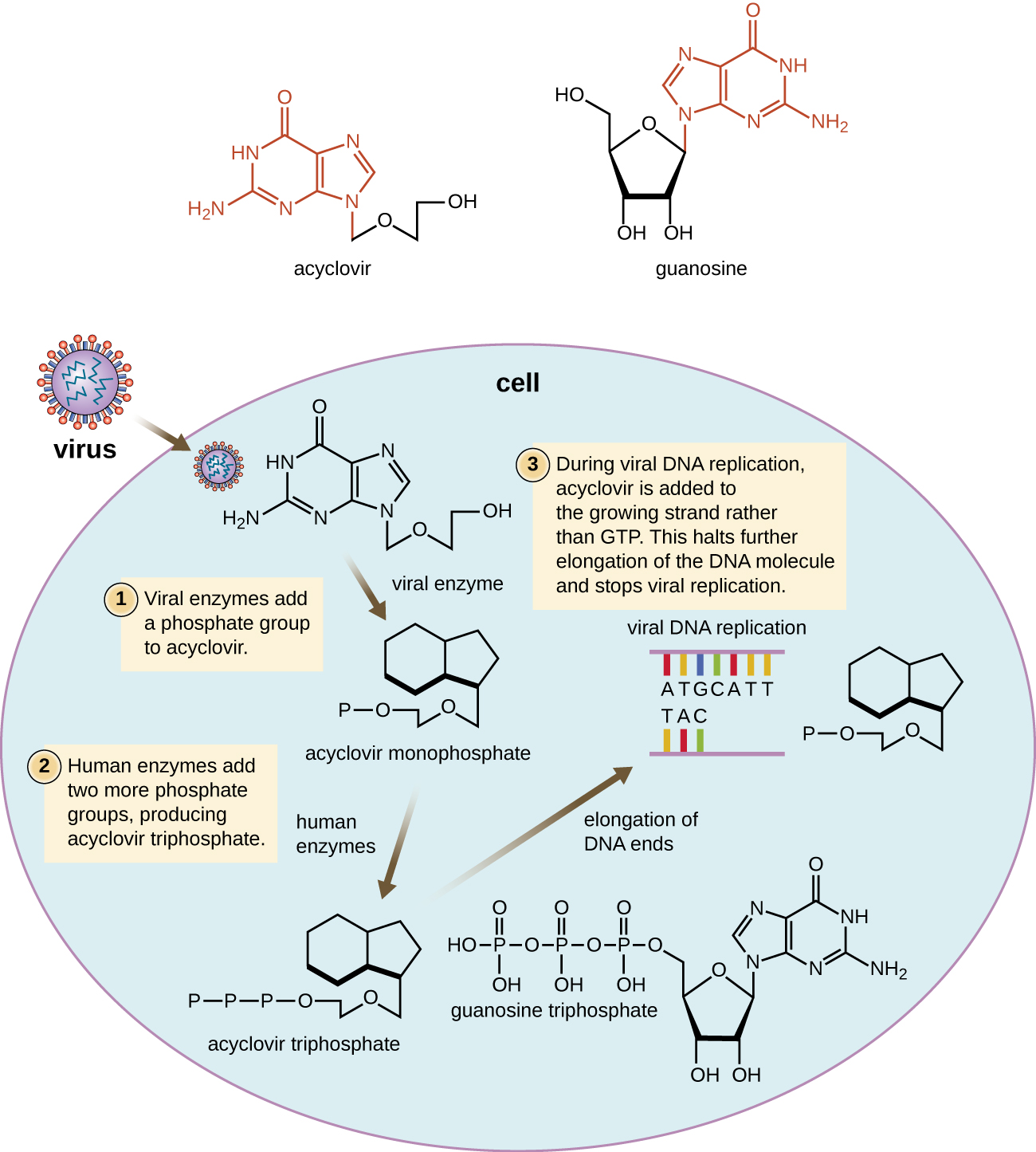
Muchos medicamentos antivirales son análogos de nucleósidos y funcionan inhibiendo la biosíntesis de ácidos nucleicos. Por ejemplo, el aciclovir (comercializado como Zovirax) es un análogo sintético del nucleósido guanosina (Figura\(\PageIndex{4}\)). Se activa por la enzima viral del herpes simple timidina quinasa y, cuando se agrega a una cadena de ADN en crecimiento durante la replicación, provoca la terminación de la cadena. Su especificidad para las células infectadas por virus proviene tanto de la necesidad de una enzima viral para activarla como de la mayor afinidad de la forma activada por la ADN polimerasa viral en comparación con la ADN polimerasa de la célula hospedadora. El aciclovir y sus derivados se utilizan frecuentemente para el tratamiento de infecciones por el virus del herpes, incluyendo herpes genital, varicela, herpes zóster, infecciones por virus de Epstein-Barr e infecciones por citomegalovirus. El aciclovir se puede administrar ya sea por vía tópica o sistémica, dependiendo de la infección. Un posible efecto secundario de su uso incluye la nefrotoxicidad. El medicamento adenina-arabinósido, comercializado como vidarabina, es un análogo sintético a la desoxiadenosina que tiene un mecanismo de acción similar al del aciclovir. También es eficaz para el tratamiento de diversos virus del herpes humano. Sin embargo, debido a los posibles efectos secundarios que involucran bajos recuentos de glóbulos blancos y neurotoxicidad, ahora se prefiere el tratamiento con aciclovir.
La ribavirina, otro análogo sintético de guanosina, funciona por un mecanismo de acción que no está del todo claro. Parece interferir tanto con la síntesis de ADN como de ARN, tal vez al reducir las reservas intracelulares de trifosfato de guanosina (GTP). La ribavarina también parece inhibir la ARN polimerasa del virus de la hepatitis C. Se utiliza principalmente para el tratamiento de los virus ARN como la hepatitis C (en terapia combinada con interferón) y el virus sincitial respiratorio. Los posibles efectos secundarios del uso de ribavirina incluyen anemia y efectos sobre el desarrollo en niños por nacer en pacientes embarazadas. En los últimos años, también se ha desarrollado otro análogo nucleotídico, el sofosbuvir (Solvaldi), para el tratamiento de la hepatitis C. El sofosbuvir es un análogo de uridina que interfiere con la actividad de la polimerasa viral. Comúnmente se coadministra con ribavirina, con y sin interferón.
La inhibición de la síntesis de ácidos nucleicos no es el único objetivo de los antivirales sintéticos. Si bien el modo de acción de la amantadina y su relativa rimantadina no están del todo claros, estos fármacos parecen unirse a una proteína transmembrana que está implicada en el escape del virus de la influenza de los endosomas. El bloqueo del escape del virus también evita la liberación de ARN viral en las células hospedadoras y la posterior replicación viral. El aumento de la resistencia ha limitado el uso de amantadina y rimantadina en el tratamiento de la influenza A. El uso de amantadina puede provocar efectos secundarios neurológicos, pero los efectos secundarios de la rimantadina parecen menos graves. Curiosamente, debido a sus efectos sobre sustancias químicas cerebrales como la dopamina y el NMDA (N-metil D-aspartato), la amantadina y la rimantadina también se utilizan para el tratamiento de la enfermedad de Parkinson.
Los inhibidores de la neuraminidasa, incluyendo olsetamivir (Tamiflu), zanamivir (Relenza) y peramivir (Rapivab), se dirigen específicamente a los virus de la influenza bloqueando la actividad de la neuraminidasa del virus de la influenza, impidiendo la liberación del virus de las células infectadas. Estos tres antivirales pueden disminuir los síntomas de la gripe y acortar la duración de la enfermedad, pero difieren en sus modos de administración: olsetamivir se administra por vía oral, zanamivir se inhala y peramivir se administra por vía intravenosa. La resistencia a estos inhibidores de la neuraminidasa todavía parece ser mínima.
El pleconaril es un antiviral sintético en desarrollo que mostró promesa para el tratamiento de picornavirus. El uso de pleconaril para el tratamiento del resfriado común causado por rinovirus no fue aprobado por la FDA en 2002 por falta de eficacia comprobada, falta de estabilidad y asociación con menstruación irregular. Su desarrollo posterior para este propósito se detuvo en 2007. Sin embargo, aún se está investigando el pleconaril para su uso en el tratamiento de complicaciones potencialmente mortales de enterovirus, como meningitis y sepsis. También se está investigando para su uso en la erradicación global de un enterovirus específico, la poliomielitis. 6 El pleconaril parece funcionar al unirse a la cápside viral y prevenir el desrecubrimiento de partículas virales dentro de las células hospedadoras durante la infección viral.
Los virus con ciclos de vida complejos, como el VIH, pueden ser más difíciles de tratar. En primer lugar, el VIH se dirige a los glóbulos blancos CD4 positivos, que son necesarios para una respuesta inmune normal a la infección. Segundo, el VIH es un retrovirus, lo que significa que convierte su genoma de ARN en una copia de ADN que se integra en el genoma de la célula hospedadora, escondiéndose así dentro del ADN de la célula hospedadora. En tercer lugar, la transcriptasa inversa del VIH carece de actividad correctora e introduce mutaciones que permiten un rápido desarrollo de resistencia a los medicamentos antivirales. Para ayudar a prevenir la aparición de resistencia, se suele utilizar una combinación de medicamentos antivirales sintéticos específicos en el TAR para el VIH (Figura\(\PageIndex{5}\)).
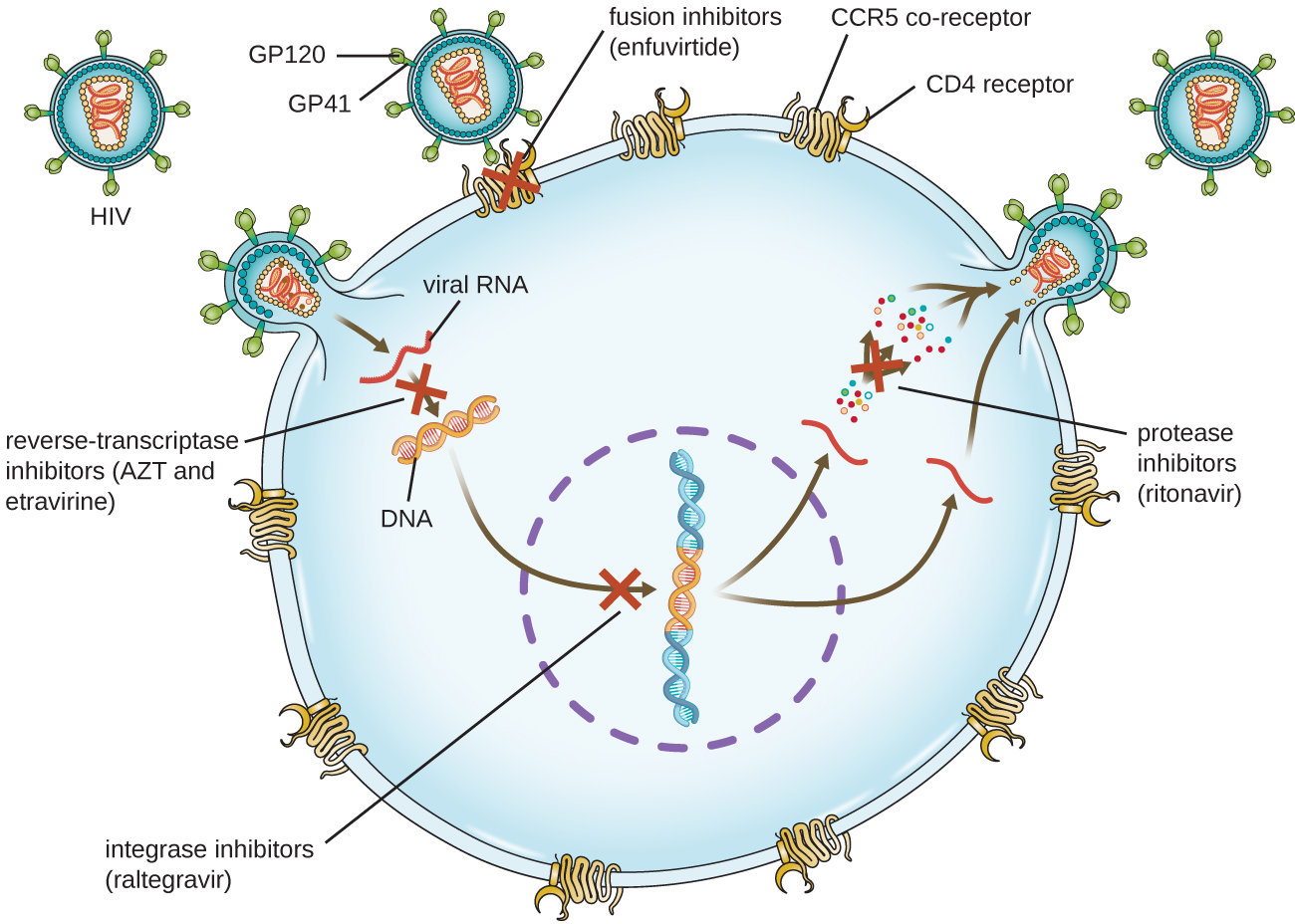
Los inhibidores de la transcriptasa inversa bloquean la etapa temprana de convertir el genoma de ARN viral en ADN, y pueden incluir inhibidores análogos de nucleósidos competitivos (por ejemplo, azidotimidina/zidovudina, o AZT) e inhibidores no competitivos no nucleósidos (por ejemplo, etravirina) que se unen a la transcriptasa inversa y causan una inactivar el cambio conformacional. Los fármacos llamados inhibidores de la proteasa (p. ej., ritonavir) bloquean el procesamiento de las proteínas virales y previenen la maduración viral. También se están desarrollando inhibidores de proteasas para el tratamiento de otros tipos virales. 7 Por ejemplo, simeprevir (Olysio) ha sido aprobado para el tratamiento de la hepatitis C y se administra con ribavirina e interferón en terapia de combinación. Los inhibidores de la integrasa (p. ej., raltegravir), bloquean la actividad de la integrasa del VIH responsable de la recombinación de una copia de ADN del genoma viral en el cromosoma de la célula huésped. Las clases de fármacos adicionales para el tratamiento del VIH incluyen los antagonistas de CCR5 y los inhibidores de fusión (por ejemplo, enfuviritida), que impiden la unión del VIH al correceptor de la célula huésped (receptor de quimiocina tipo 5 [CCR5]) y la fusión de la envoltura viral con la membrana de la célula huésped, respectivamente. En el cuadro se\(\PageIndex{4}\) muestran las diversas clases terapéuticas de medicamentos antivirales, categorizados por modo de acción, con ejemplos de cada uno.
| Mecanismo de Acción | Droga | Usos Clínicos |
|---|---|---|
| Inhibición de análogos de nucleósidos de la síntesis de ácidos nucleicos | Aciclovir | Infecciones por virus del herpes |
| Azidotimidina/zidovudina (AZT) | Infecciones por VIH | |
| Ribavirina | Infecciones por el virus de la hepatitis C y virus sincitial respiratorio | |
| Vidarabine | Infecciones por virus del herpes | |
| Sofosbuvir | Infecciones por virus de hepatitis C | |
| Inhibición no competitiva no nucleósido | Etravirina | Infecciones por VIH |
| Inhiben el escape de virus de los endosomas | Amantadina, rimantadina | Infecciones por virus de la influenza |
| Inhibe la neuraminadasa | Olsetamivir, zanamivir, peramivir | Infecciones por virus de la influenza |
| Inhibe el desrecubrimiento viral | Peconaril | Infecciones graves por enterovirus |
| Inhibición de proteasa | Ritonavir | Infecciones por VIH |
| Simeprevir | Infecciones por virus de hepatitis C | |
| Inhibición de la integrasa | Raltegravir | Infecciones por VIH |
| Inhibición de la fusión de membrana | Enfuviritida | Infecciones por VIH |
Ejercicio\(\PageIndex{4}\)
¿Por qué es difícil tratar el VIH con antivirales?
Para conocer más sobre las diversas clases de medicamentos antirretrovirales utilizados en el TAR de la infección por VIH, explore cada uno de los medicamentos de las clases de medicamentos contra el VIH proporcionadas por el Departamento de Salud y Servicios Humanos de Estados Unidos en este sitio web.
Conceptos clave y resumen
- Debido a que los hongos, protozoos y helmintos son organismos eucariotas como las células humanas, es más difícil desarrollar medicamentos antimicrobianos que se dirijan específicamente a ellos. Del mismo modo, es difícil apuntar a los virus porque los virus humanos se replican dentro de las células humanas.
- Los fármacos antifúngicos interfieren con la síntesis de ergosterol, se unen al ergosterol para alterar la integridad de la membrana celular fúngica o dirigirse a componentes específicos de la pared celular u otras proteínas celulares.
- Los fármacos antiprotozoarios aumentan los niveles celulares de especies reactivas de oxígeno, interfieren con la replicación del ADN protozoario (nuclear versus ADNk, respectivamente) e interrumpen la desintoxicación del hemo.
- Los fármacos antihelmínticos interrumpen la formación de microtúbulos helmínticos y protozoarios; bloquean las transmisiones neuronales; inhiben la formación anaeróbica de ATP y/o la fosforilación oxidativa; inducen una afluencia de calcio en las tenias, provocando espasmos y parálisis; e interfieren con la síntesis de ARN en esquistosomas.
- Los medicamentos antivirales inhiben la entrada viral, inhiben el desrecubrimiento viral, inhiben la biosíntesis de ácidos nucleicos, previenen el escape viral de los endosomas en las células hospedadoras y previenen la liberación viral de las células infectadas.
- Debido a que puede mutar fácilmente para volverse resistente a los medicamentos, el VIH generalmente se trata con una combinación de varios medicamentos antirretrovirales, que pueden incluir inhibidores de la transcriptasa inversa, inhibidores de proteasa, inhibidores de la integrasa y fármacos que interfieren con la unión viral y la fusión a iniciar la infección.
Notas al pie
- 1 Centros de Control y Prevención de Enfermedades. “Fiebre del Valle: La Conciencia Es Clave.” www.cdc.gov/features/valleyfever/. Accedido el 1 de junio de 2016.
- 2 B. Chu et al. “Un derivado de bencimidazol que exhibe actividad antitumoral bloquea la actividad de EGFR y HER2 y regula positivamente DR5 en células de cáncer de mama”. Muerte celular y enfermedad 6 (2015) :e1686
- 3 J.-X. Pan et al. “La niclosamida, un viejo agente antihelmíntico, demuestra actividad antitumoral al bloquear múltiples vías de señalización de células madre cancerosas”. Revista china del cáncer 31 núm. 4 (2012) :178—184.
- 4 F. Imperi et al. “Nueva vida para una droga vieja: El medicamento antihelmíntico La niclosamida inhibe la detección de quórum de Pseudomonas aeruginosa”. Agentes Antimicrobianos y Quimioterapia 57 núm. 2 (2013) :996-1005.
- 5 A. Jurgeit et al. “La niclosamida es un portador de protones y se dirige a los endosomas ácidos con amplios efectos antivirales”. PLoS Patógenos 8 núm. 10 (2012) :e1002976.
- 6 M.J. Abzug. “Los Enterovirus: Problemas en Necesidad de Tratamientos”. Diario de Infección 68 no. S1 (2014) :108—14.
- 7 B.L. Pearlman. “Inhibidores de Proteasas para el Tratamiento de la Infección Crónica de Hepatitis C Genotipo-1: El Nuevo Estándar de Atención”. Lancet Enfermedades Infecciosas 12 núm. 9 (2012) :717—728.