
2.4: Enfermedades Neurodegenerativas Crónicas
- Page ID
- 121864

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Alfa-sinucleinopatías y enfermedad de Parkinson
El modelo actual para la mayoría de las condiciones neurodegenerativas es que las neuronas mueren a través de la apoptosis o una forma programada de muerte celular. Esto tiene implicaciones clínicas muy importantes ya que activar o desactivar genes/proteínas específicos es un método para detener o posiblemente revertir la neurodegeneración. Se cree que las enfermedades neurodegenerativas como la enfermedad de Parkinson y la enfermedad de Alzheimer y otras utilizan vías apoptóticas y, además, sugieren que hay una proteína componente que se pliega aberrantemente para producir apoptosis.
Enfermedad de Parkinson: Resumen
La mayoría de los casos de este trastorno parecen ser esporádicos (es decir, de origen no genético). Muy pocos casos parecen tener un origen genético (por ejemplo, los genes más frecuentemente asociados con los genes DJ-1, Parkin (ligasa Ubiquitina E3) y alfa sinucleína). La enfermedad de Parkinson se asocia con mayor frecuencia con la pérdida de núcleos “pigmentados” en el cerebro y típicamente implica la pérdida de un grupo de neuronas que se encuentran en la Sustancia Negra (Figura\(\PageIndex{1}\).). Las neuronas de la sustancia negra son dopaminérgicas y pigmentadas porque contienen la proteína melanina.
El inicio típico de la enfermedad de Parkinson es de etapas medias a posteriores de la vida (es decir, 50 y más) aunque un pequeño porcentaje de individuos que han conocido mutaciones genéticas en Parkin o alfa-sinucleína desarrollan síntomas antes.
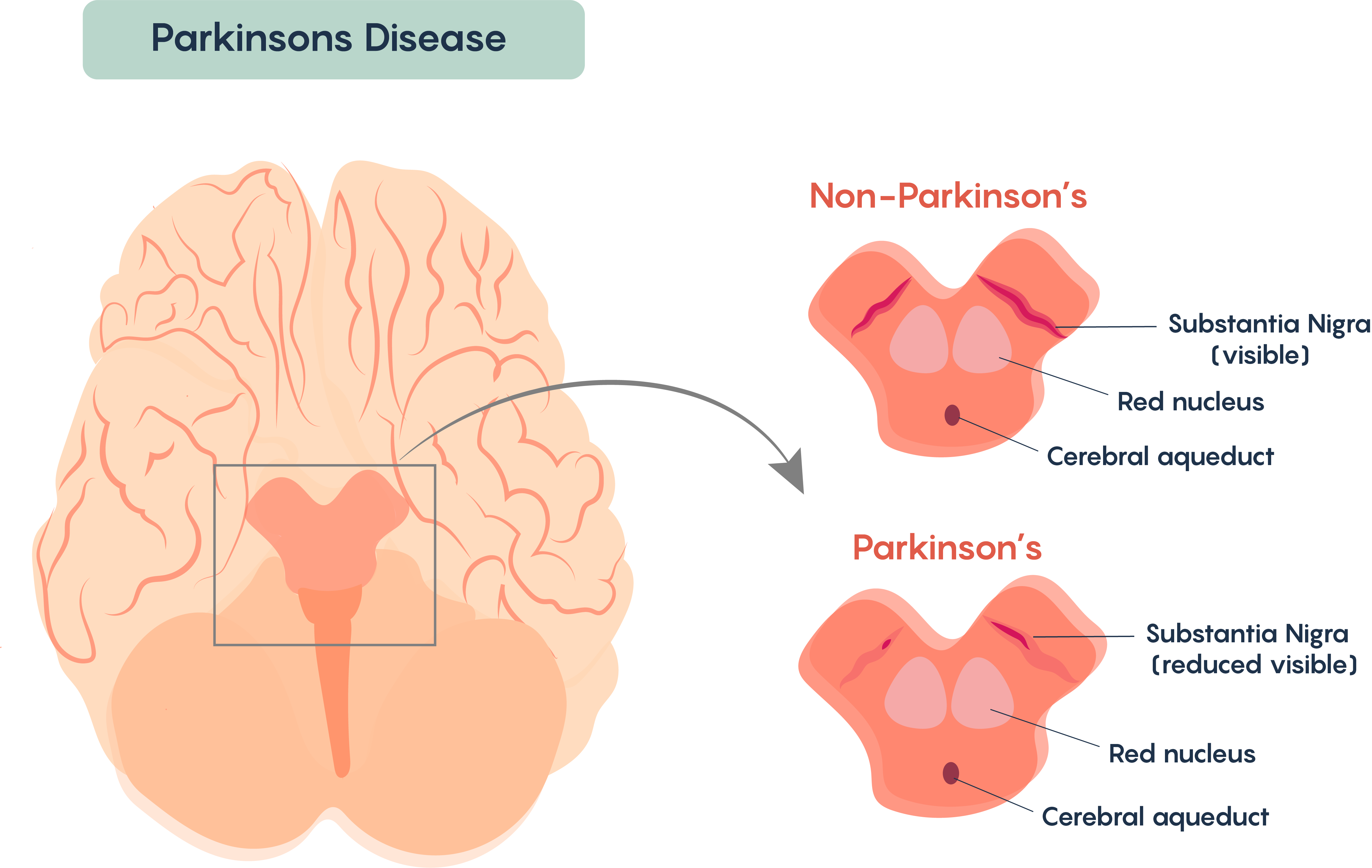
Figura\(\PageIndex{1}\). Pérdida de las neuronas dopaminérgicas pigmentadas dentro de la Substancia Nigra de individuos con Enfermedad de Parkinson.
La pérdida de estas células afecta el procesamiento y ejecución del movimiento voluntario en individuos con Enfermedad de Parkinson. Similar a la esclerosis múltiple, una vez diagnosticados, los síntomas se vuelven continuos y progresivos, es decir, los síntomas empeoran con el tiempo. Nuevamente, hay muchas similitudes con la Esclerosis Múltiple, y no se conoce una cura para la Enfermedad de Parkinson y la enfermedad sigue siendo idiopática aunque hay algunas causas conocidas de la Enfermedad de Parkinson, incluyendo pérdida de movimiento tras aterosclerosis cerebral, encefalitis viral, y como resultado de efectos de fármacos como fenotiazidas y reserpina.
Síntomas “clásicos” asociados a la enfermedad de Parkinson
- Bradicinesia: lentitud en la iniciación y ejecución de movimientos voluntarios
- Rigidez: Aumenta el tono muscular y aumenta la resistencia al movimiento (rigidez de brazos y piernas), a medida que aumenta la severidad produce rigidez de rueda dentada
- Temblor: Por lo general, temblor en reposo; Cuando la persona se sienta, el brazo tiembla; Temblor se detiene cuando la persona intenta agarrar algo (temblor rodante de pastillas)
- Inestabilidad postural: fijación anormal de la postura (agacharse al estar de pie), problemas con el equilibrio y reflejo de corrección
- Alteración de la marcha: barajar pies
- Hipotensión ortostática
- Demencia (en algunos casos)
- Distonía (contracción muscular inapropiada y continua)
- Oftalmoplejía (debilidad en los músculos oculares)
- Trastornos afectivos del estado de ánimo (como depresión mayor)
Cuerpos de Lewy y alfa-sinucleína: características distintivas de la enfermedad de Parkinson
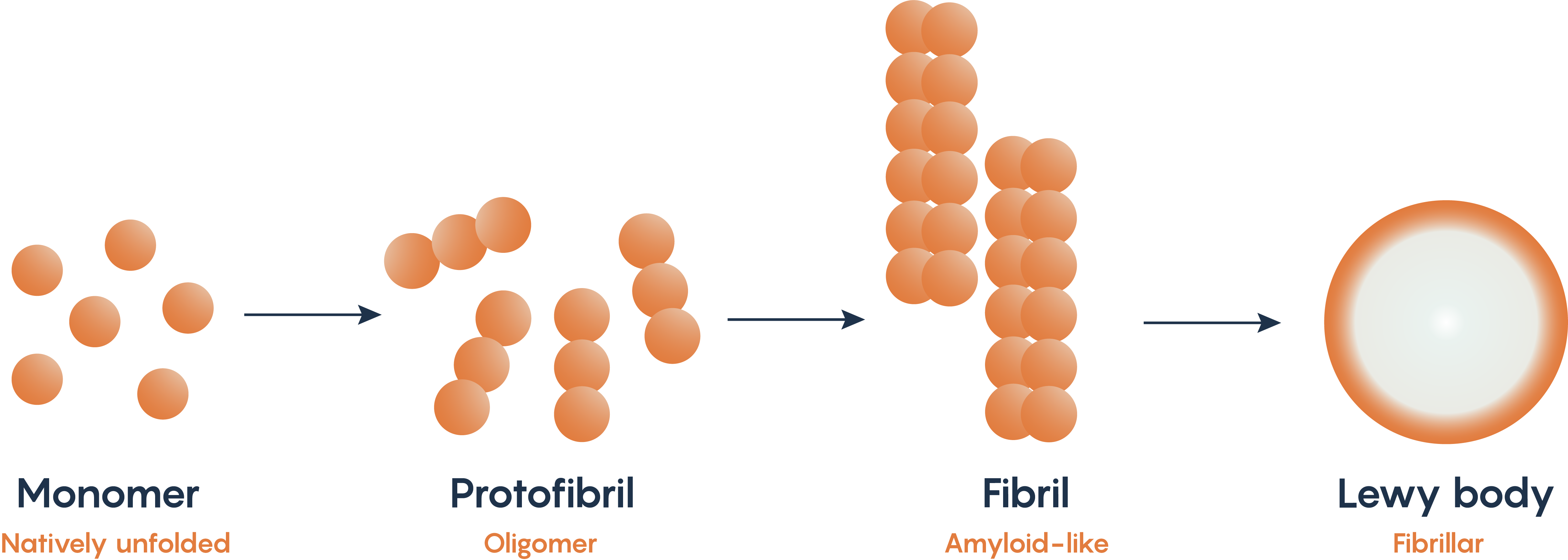
Figura\(\PageIndex{2}\). Desplegamiento y posterior acumulación de alfa-sinucleína.
La alfa-sinucleína es una proteína natural dentro de las neuronas. Las mutaciones en los genes PARK1 y PARK4 que normalmente codifican para alfa-sinucleína, se han asociado con la enfermedad de Parkinson. Como tal, muchos modelos animales de la Enfermedad de Parkinson buscan la producción de la forma fibrilar de la alfa-sinucleína a medida que se despliega y luego se acumula dentro de las neuronas de la sustancia nigral conocidas como cuerpos de Lewy (Figura\(\PageIndex{2}\).). Se ha planteado la hipótesis de que el mal plegamiento y acumulación de alfa-sinucleína es la razón por la que las neuronas sufren apoptosis, aunque el mecanismo exacto de cómo ocurre esto aún no se ha aclarado.
Modelos animales de la enfermedad de Parkinson
La falta de genes candidatos (a excepción de alfa-sinucleína, Parkin y DJ-1) ha significado que la mayoría de los científicos hayan mirado modelos de toxinas. La mayoría de las toxinas que producen características que se asemejan a cambios en el movimiento, así como a la formación de cuerpos de Lewy usan destrucción catecolaminérgica. De hecho, la mayoría de estas toxinas son inhibidores del complejo mitocondrial increíblemente poderosos y peligrosos como reserpina, MPTP, metanfetamina, 6-OH-dopamina, rotenona y paraquat). De hecho, una toxina, el MPTP es un subproducto de la producción de heroína sintética, lo que sugiere que puede haber una sustancia sintética que cause la enfermedad de Parkinson y algunos estudios epidemiológicos muestran una correlación con el uso de pesticidas (como paraquat y rotenona) y la enfermedad de Parkinson.
Enfermedad de Alzheimer: visión general y patología general
Otro padecimiento neurodegenerativo es la Enfermedad de Alzheimer (EA). Esta afección neurodegenerativa es una forma de demencia caracterizada por los síntomas más comunes asociados a la enfermedad de Alzheimer notablemente pérdida de memoria, problemas de comunicación y dificultad para encontrar palabras, problemas de atención, confusión y desorientación espacial, disminución o mal juicio, cambios en el estado de ánimo y personalidad.
Las áreas afectadas explican la patología de la EA, que se caracteriza por la pérdida de memoria debido a la contracción del hipocampo, y otros problemas que incluyen el pensamiento y el rendimiento de mayor nivel que son controlados por la corteza. La atrofia cerebral generalmente comienza en el lóbulo temporal medial (es decir, el hipocampo), pasa a las córtices de asociación y, por lo tanto, afecta las áreas sensoriales y motoras. También afecta al Núcleo Basalis de Meynert que tiene múltiples proyecciones colinérgicas a la corteza (y se cree que es responsable del control del sueño, la atención y la conciencia) como se destaca en la Figura\(\PageIndex{3}\).
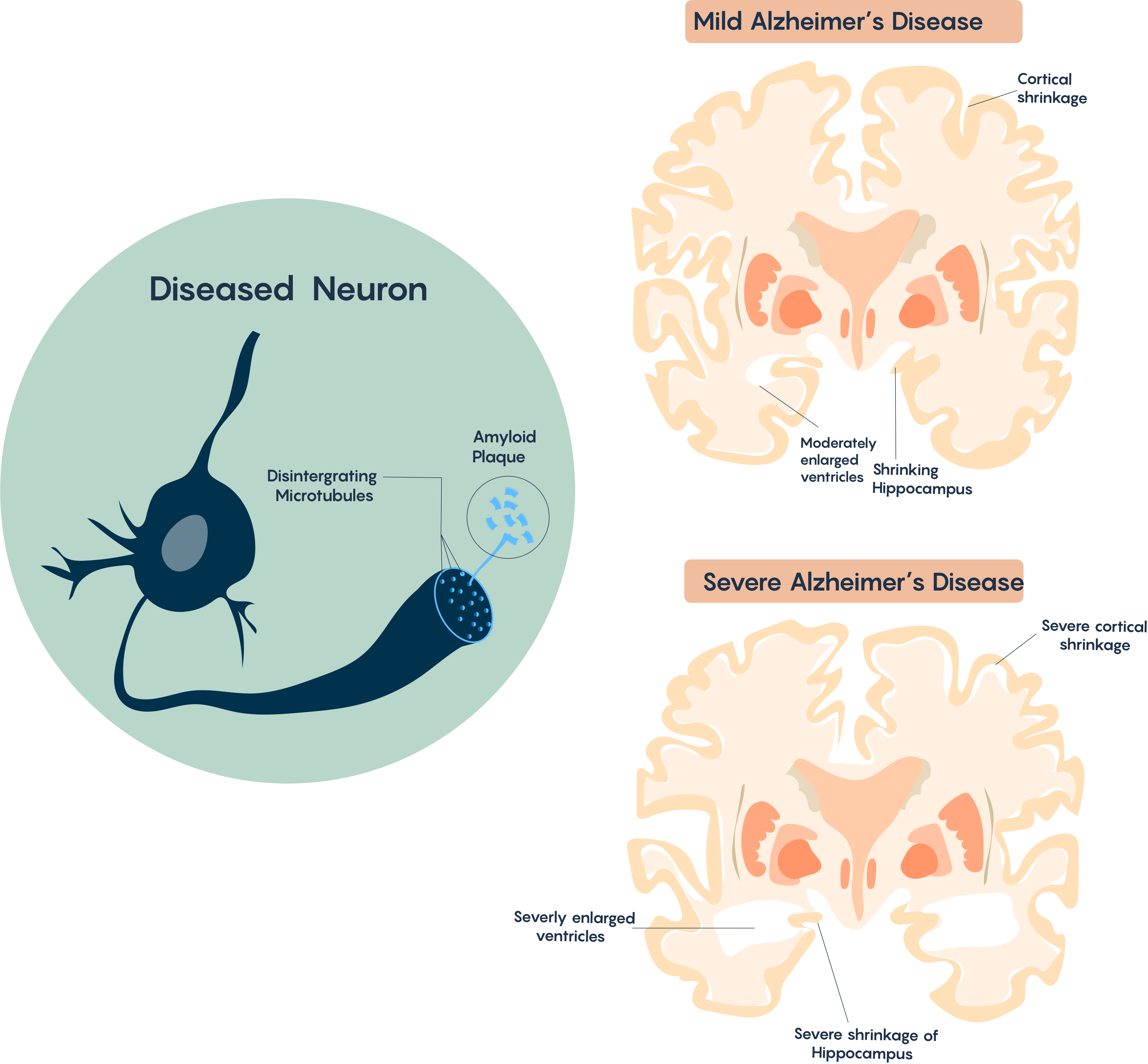
Figura\(\PageIndex{3}\). Contracción y pérdida de regiones específicas del cerebro.
Patología Celular
El cerebro típico de la EA se caracteriza por la pérdida de neuronas, pérdida progresiva de sinapsis y proyecciones colinérgicas, la acumulación de placas extracelulares β-amiloides (Aβ) y oidos neurofibrilares intracelulares (NFTs). Con mayor tiempo y patología esto a menudo se correlacionará con la formación de una cicatriz glial (involucrando astrocitos reactivos). También hay evidencia muy fuerte de que este trastorno también puede tener un fuerte componente inmune ya que a menudo existe la infiltración de células microgliales en el cerebro de la Enfermedad de Alzheimer.
Placas β-amiloides (Aβ): Hipótesis de βA Cascada
Una de las características histopatológicas y moleculares de la enfermedad de Alzheimer es la presencia de agregados extracelulares que incluyen placas amiloides. Se cree que estas placas inducen citotoxicidad al alterar las funciones neuronales normales, las concentraciones de iones y la generación de potencial de acción. Al igual que con la teoría moderna de la neurodegeneración, estas placas están compuestas por la forma escindida de la proteína precursora amiloide (APP).
APP se puede procesar a través de dos vías diferentes:
- Vía no amiloidogénica (escindida por α- y γ-secretasas)
- Vía amiloidogénica (escindida por β- y γ-secretasas)
Procesamiento de APP
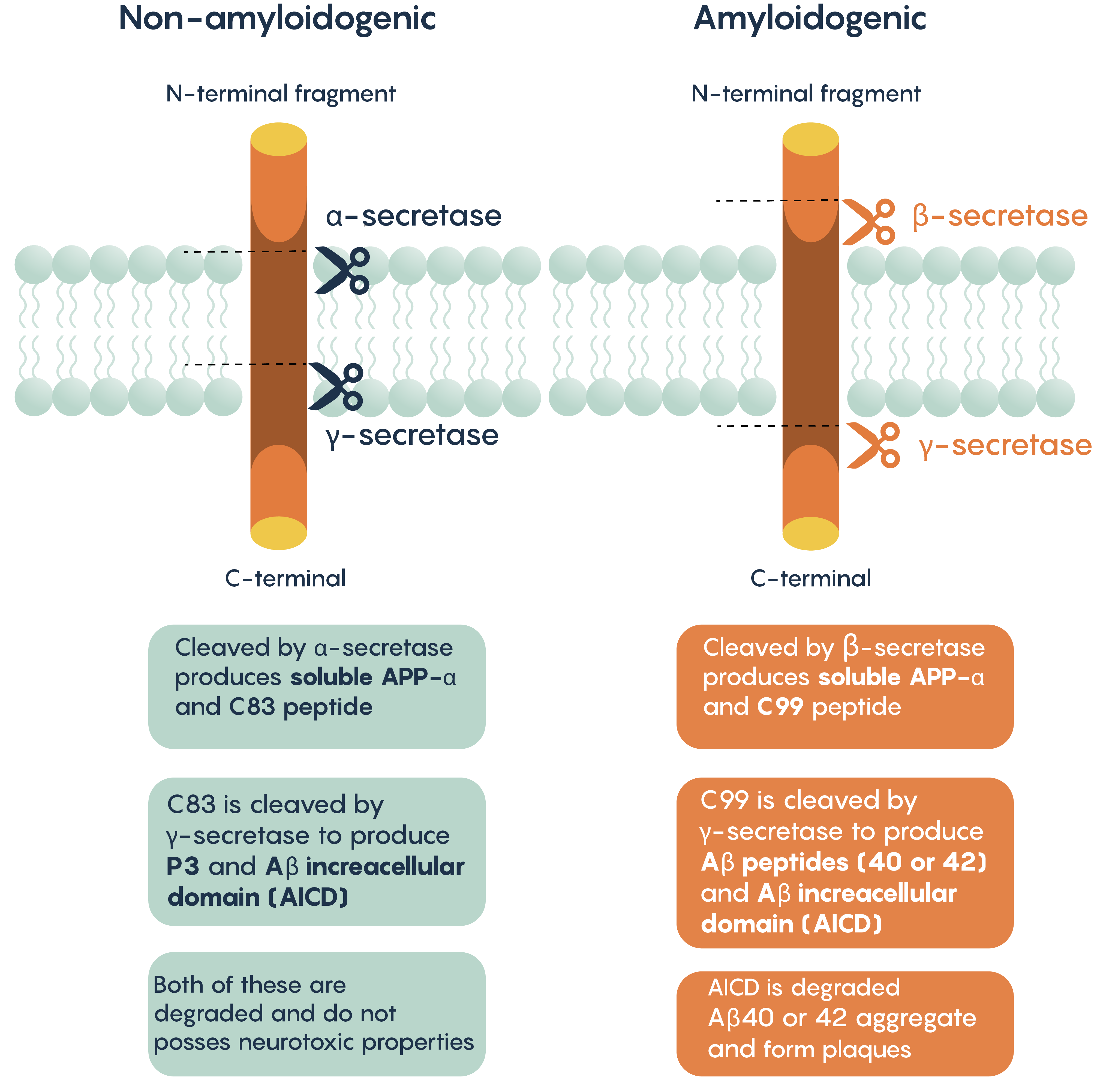
Figura\(\PageIndex{4}\). Vías de procesamiento de APP que conducen a la producción y agregación de β-amiloide.
Enredos neurofibrilares (NFTs)
Otra característica del cerebro enfermo de Alzheimer es la aparición de enredos neurofibrilares (NFTs) que están compuestos por proteína tau hiperfosforilada. Normalmente dentro de las neuronas (y de hecho la mayoría de las células), la tau se une y estabiliza los microtúbulos (MTs), sin embargo, cuando ésta es fosforilada por las quinasas, la tau pierde su afinidad por las MTs y se disocia del complejo MT. Esto hace que los microtúbulos a su vez se desensamblen lo que interfiere con el transporte axonal adecuado y eventualmente conduce a la pérdida de integridad neuronal. Como tal, las proteínas fosfo-Tau forman agregados intracelulares (NFTs) que se convierten en otra característica definitoria de esta enfermedad, así como una diana potencial para la intervención terapéutica.
Genética de la Enfermedad de Alzheimer
Mientras que los casos esporádicos de EA representan la mayoría de los pacientes con EA, la EA familiar (FAD) representa solo el 5% de ellos. La forma familiar, FAD se manifiesta antes (alrededor de 40-50 años de edad) en comparación con >65 años de edad en casos esporádicos. A pesar de esta drástica diferencia en el inicio de la enfermedad, los síntomas son idénticos. Solo el 50% de los casos de FAD pueden explicarse por mutaciones conocidas en genes que codifican APP y presenilinas 1 y 2.
Las mutaciones de APP son relativamente raras y se caracterizan por individuos que presentan síntomas con un inicio promedio de edad a principios de los 50 años; la conclusión de que las mutaciones APP podrían causar la enfermedad de Alzheimer se basa en la observación de que la mayoría de los pacientes con síndrome de Down también desarrollan EA después de los 40 años ya que tienen un extra copia de Ch21 (que codifica para APP). Sin embargo, la mayoría de las formas heredadas de mutaciones APP alteran el procesamiento de APP
- aumentar la escisión a través de la vía de la β-secretasa
- aumentar la relación Aβ42/40
- por lo tanto, conducen a la producción de péptidos con mayor potencial fibrilogénico (Figura\(\PageIndex{2}\).)