
5.2: Asamblea Genómica I- Superposición-Diseño-Enfoque de Consenso
- Page ID
- 54096

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Muchas áreas de investigación en biología computacional se basan en la disponibilidad de datos completos de la secuencia del genoma completo. Sin embargo, el proceso para secuenciar un genoma completo no es trivial y un área de investigación activa. El problema radica en el hecho de que las tecnologías actuales de secuenciación genómica no pueden leer continuamente de un extremo de una larga secuencia genómica al otro; solo pueden secuenciar con precisión pequeñas secciones de pares de bases (que van de 100 a unos pocos miles, dependiendo del método), llamadas lecturas. Por lo tanto, para construir una secuencia de millones o miles de millones de pares de bases (como el genoma humano), los biólogos computacionales deben encontrar formas de combinar lecturas más pequeñas en secuencias de ADN continuas más grandes. Primero, examinaremos aspectos de la configuración experimental para el enfoque de superposición, diseño y consenso, y luego avanzaremos hacia el aprendizaje sobre cómo combinar lecturas y aprender información de ellas
Configuración del experimento
El primer reto que se debe abordar a la hora de poner en marcha este experimento es que necesitamos comenzar con muchas copias de cada cromosoma para poder utilizar este enfoque. Este número es del orden de 10 5. Es importante señalar que la forma en que obtengamos estas copias es muy importante y afectará nuestros resultados más adelante ya que muchas de las comparaciones que hagamos dependerán de datos consistentes. La primera forma en que podemos pensar para obtener tantos datos es amplificar un genoma dado. Sin embargo, la amplificación hace daño lo que arrojará nuestros algoritmos en pasos posteriores y causará peores resultados. Otro método posible sería congrudear el genoma para obtener muchas copias de cada cromosoma. Si buscas deshacerte del polimorfismo, esta puede ser una buena técnica, pero también perdemos datos valiosos de los sitios polimórficos cuando nos cruzamos. Un método sugerido para obtener estos datos es usar un individuo, aunque el organismo tendría que ser bastante grande. También podríamos usar técnicas como la progenie de uno o progenie de dos para obtener la menor cantidad posible de versiones de cada cromosoma. Esto conseguirá una alta profundidad de secuenciación en cada cromosoma, que es la razón por la que queremos que todos los cromosomas sean lo más similares posible.
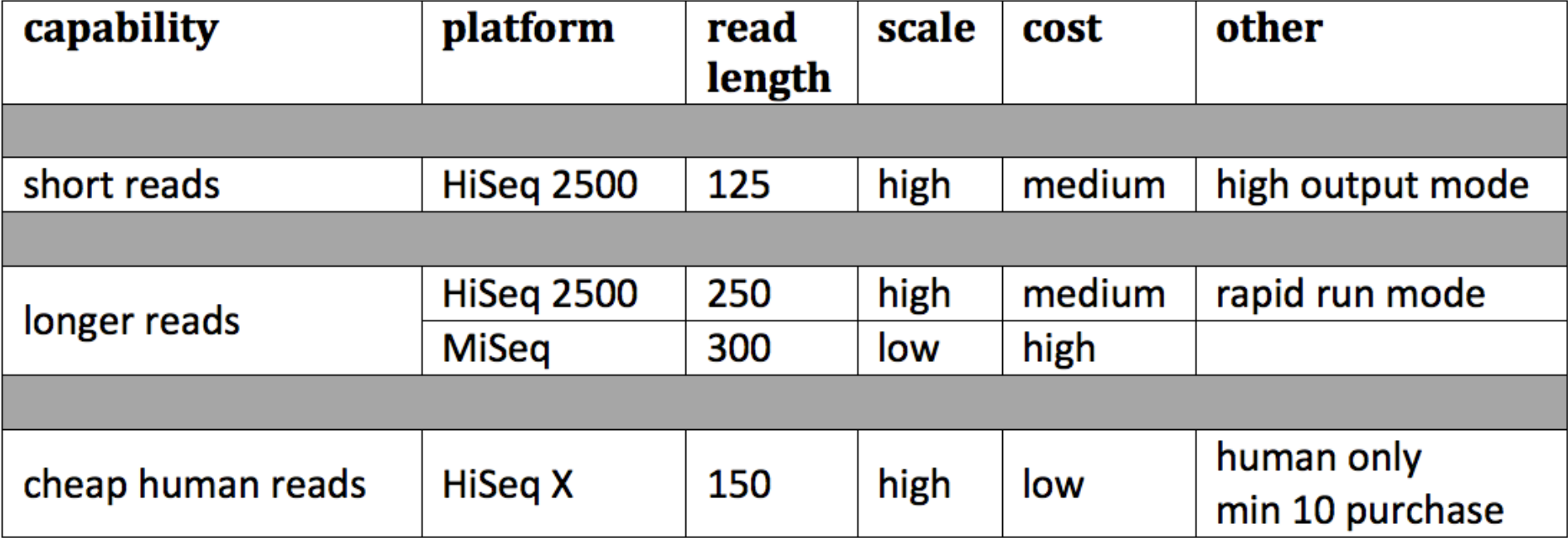
A continuación, veamos cómo podríamos decidir sobre nuestras longitudes de lectura dada la tecnología actual. Mirando (Figura 5.2), podemos ver que se debe hacer un análisis costo-beneficio para decidir qué plataforma usar en un proyecto determinado. Con la tecnología actual, comúnmente usamos HiSeq2500 con una longitud de lectura de aproximadamente 250, aunque esto está cambiando rápidamente.
Por último, veamos algunas secuencias que causan problemas al usar plataformas con lecturas cortas. Las secuencias con alto contenido de GC (por ejemplo, GGCGCGATC), bajo contenido de GC (por ejemplo, AAATAATCAA) o baja complejidad (por ejemplo, ATATATA) pueden causar problemas con lecturas cortas. Esta sigue siendo un área activa de investigación, pero algunas explicaciones posibles incluyen el deslizamiento de la polimerasa y la desnaturalización del ADN con demasiada facilidad o no lo suficientemente fácil.
En esta sección se examinará uno de los métodos tempranos más exitosos para ensamblar computacionalmente un genoma a partir de un conjunto de lecturas de ADN, llamado secuenciación de escopeta (Figura 5.3). La secuenciación de escopeta implica cortar aleatoriamente múltiples copias del mismo genoma en muchos fragmentos pequeños, como si el ADN fuera disparado con una escopeta. Típicamente, el ADN se fragmenta realmente usando sonicación (breves ráfagas de un ultrasonido) o una enzima dirigida diseñada para escindir el genoma en motivos de secuencia específicos. Ambos métodos se pueden ajustar para crear fragmentos de diferentes tamaños.
Después de que el ADN ha sido amplificado y fragmentado, se utiliza la técnica desarrollada por Frederick Sanger en 1977 llamada secuenciación de terminación de cadena (también llamada secuenciación de Sanger) para secuenciar los fragmentos. En resumen, los fragmentos son extendidos por la ADN polimerasa hasta que se incorpora un didesoxinucleotrifosfato; estos nucleótidos especiales provocan la terminación de la extensión de un fragmento. Por lo tanto, la longitud del fragmento se convierte en un proxy para donde se agregó un ddNTP dado en la secuencia. Se pueden ejecutar cuatro reacciones separadas, cada una con un ddNTP diferente (A, G, C, T) y luego ejecutar los resultados en un gel para determinar el orden relativo de las bases. El resultado son muchas secuencias de bases con puntuaciones de calidad por base correspondientes, lo que indica la probabilidad de que cada base haya sido llamada correctamente. Los fragmentos más cortos se pueden secuenciar completamente, pero los fragmentos más largos solo se pueden secuenciar en cada uno de sus extremos ya que la calidad disminuye significativamente
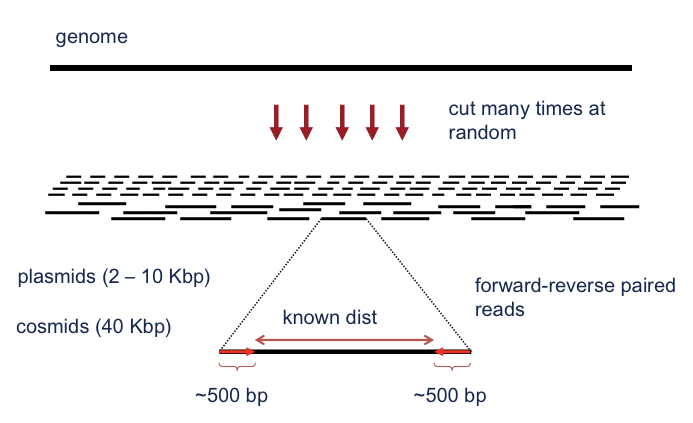
Figura 5.3: La secuenciación de escopeta implica cortar aleatoriamente un genoma en pequeños fragmentos para que puedan ser secuenciados y luego reensamblarlos computacionalmente en una secuencia continua.
después de aproximadamente 500-900 pares de bases. Estas lecturas de extremos pareados se denominan pares de relaciones de pareja. En el resto de esta sección, discutimos cómo usar las lecturas para construir secuencias mucho más largas, hasta el tamaño de cromosomas completos.
Encontrar lecturas superpuestas
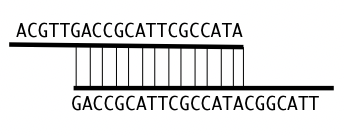
Para combinar los fragmentos de ADN en segmentos más grandes, debemos encontrar lugares donde dos o más lecturas sobre- lap, es decir, donde la secuencia inicial de un fragmento coincide con la secuencia final de otro fragmento. Por ejemplo, dados dos fragmentos como ACGTTGACCGCATTCGCCATA y GACCGCATTCGCCATACG- GCATT, podemos construir una secuencia mayor basada en el solapamiento: ACGTTGACCGCATTCGCCATACGCATGCCATACGCATT (Figura 5.4).
Un método para encontrar secuencias coincidentes es el algoritmo de programación dinámica de Needleman-Wunsch, que se discutió en el capítulo 2. El método Needleman-Wunsch no es práctico para el ensamblaje del genoma, sin embargo, ya que necesitaríamos realizar millones de alineamientos de pares, cada uno tomando O (n 2) tiempo, para construir un genoma completo a partir de los fragmentos de ADN.
Un mejor enfoque es usar el algoritmo BLAST (discutido en el capítulo 3) para hash todos los k-meros (secuencias únicas de longitud k) en las lecturas y encontrar todas las ubicaciones donde dos o más lecturas tienen uno de los k-meros en común. Esto nos permite lograr O (k n) eficiencia en lugar de O (n 2) comparaciones por parejas. k puede ser cualquier número menor que el tamaño de las lecturas, pero varía dependiendo de la sensibilidad y especificidad deseadas. Ajustando la longitud de lectura para abarcar las regiones repetitivas del genoma, podemos resolver correctamente estas regiones y acercarnos mucho al ideal de un genoma completo y continuo. Un ensamblador popular de superposición-diseño-consenso llamado Arachne usa k = 24 [2].
Dados los k-mers coincidentes, podemos alinear cada una de las lecturas correspondientes y descartar cualquier coincidencia que sea inferior al 97% similar. No requerimos que las lecturas sean idénticas ya que permitimos la posibilidad de errores de secuenciación y heterocigosidad (es decir, un organismo diploide como un ser humano puede tener dos variantes diferentes en un sitio polimórfico).
Fusionando lecturas en cóntigs
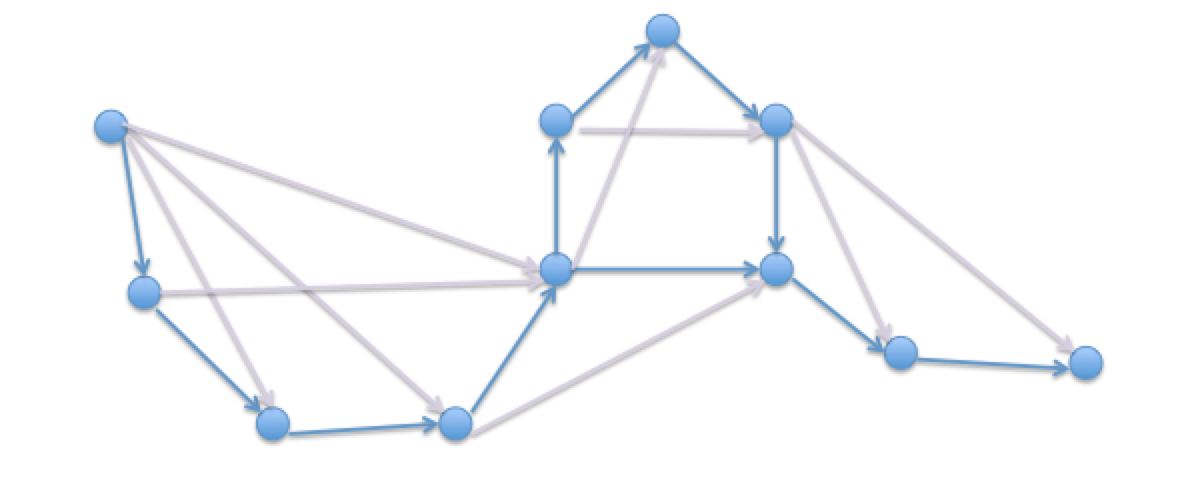
Utilizando las técnicas descritas anteriormente para encontrar superposiciones entre fragmentos de ADN, podemos juntar segmentos más grandes de secuencias continuas llamadas cóntigos. Una forma de visualizar este proceso es crear una gráfica en la que todos los nodos representen lecturas, y los bordes representan superposiciones entre las lecturas (Figura 5.5). Nuestra gráfica tendrá superposición transitiva; es decir, algunos bordes conectarán nodos dispares que ya están conectados por nodos intermedios. Al eliminar las superposiciones transitoriamente inferidas, podemos crear una cadena de lecturas que han sido ordenadas para formar un cóntig más grande. Estas transformaciones gráficas se discuten con mayor profundidad en la sección 5.3.1 a continuación. Para entender mejor el tamaño de los cóntigos, calculamos algo conocido como N50. Debido a que las medidas de longitud del cóntigo tienden a ser altamente sensibles al corte del cóntigo más pequeño, N50 se calcula como la mediana ponderada por longitud. Para un ser humano, N50 suele estar cerca de 125 kb.
En teoría, deberíamos poder utilizar el enfoque anterior para crear grandes cóntigos a partir de nuestras lecturas siempre y cuando tengamos una cobertura adecuada de la región dada. En la práctica, a menudo nos encontramos con grandes secciones del genoma que son extremadamente repetitivas y como resultado son difíciles de ensamblar. Por ejemplo, no está claro exactamente cómo alinear las dos secuencias siguientes: ATATAT y ATATATAT. Debido al contenido de información extremadamente bajo en el patrón de secuencia, podrían superponerse en cualquier número de formas. Además, estas regiones repetitivas pueden aparecer en múltiples localizaciones del genoma, y es difícil determinar qué lecturas provienen de qué ubicaciones. Los cóntigos formados por estas lecturas ambiguas y repetitivas se denominan cóntigos sobrecolapsados.
Para determinar qué secciones están sobrecolapsadas, a menudo es posible cuantificar la profundidad de cobertura de los fragmentos que componen cada cóntig. Si un cóntigo tiene significativamente más cobertura que los otros, es probable que sea un candidato para una región sobrecolapsada. Adicionalmente, varios cóntigos únicos pueden superponerse a un cóntig en la misma ubicación, lo que es otra indicación de que el cóntigo puede estar sobrecolapsado (Figura 5.6).
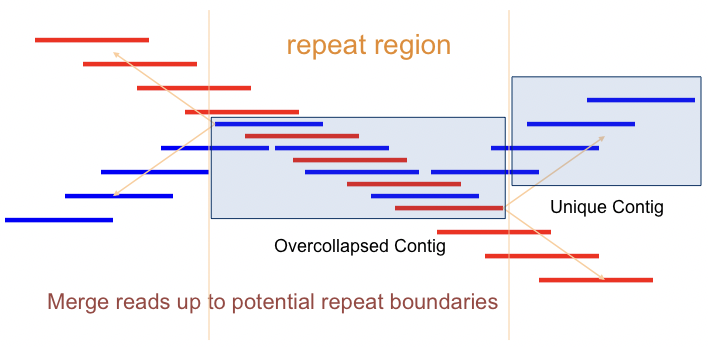
Figura 5.6: Los cóntigos sobrecolapsados son causados por regiones repetetivas del genoma que no se pueden extraer entre sí durante la secuenciación. Los patrones de ramificación de alineación que surgen durante el proceso de fusión de fragmentos en cóntigos son una fuerte indicación de que una de las regiones puede estar sobrecolapsada.
Después de que los fragmentos han sido ensamblados en cóntigos hasta el punto de una posible sección repetida, el resultado es una gráfica en la que los nodos son cóntigos, y los bordes son enlaces entre cóntigos únicos y cóntigos sobrecolapsados (Figura 5.7).
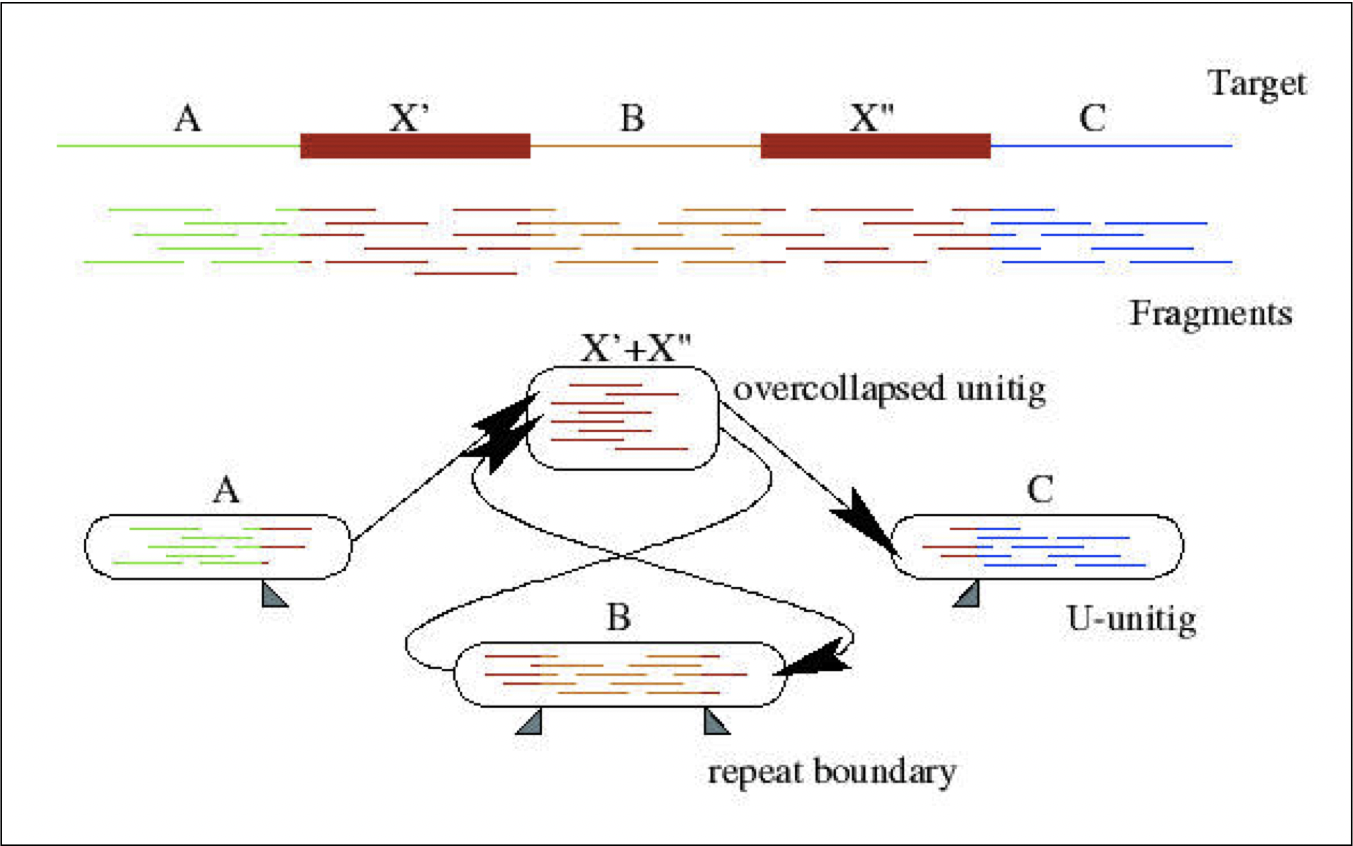
Figura 5.7: En esta gráfica que conecta cóntigos, la región repetida X tiene indegree y outdegree igual a 2. La secuencia objetivo que se muestra en la parte superior se puede inferir a partir de los enlaces en la gráfica.
Colocación de gráficos de cóntigos en andamios
Una vez que nuestros fragmentos se ensamblan en cóntigos y gráficos de cóntigos, podemos usar los pares de relaciones de pareja más grandes para unir cóntigos en supercontigs o andamios. Los pares de mate son útiles tanto para orientar los cóntigos como para colocarlos en el orden correcto. Si los pares de relaciones son lo suficientemente largos, a menudo pueden abarcar regiones repetitivas y ayudar a resolver las ambigüedades descritas en la sección anterior (Figura 5.8).
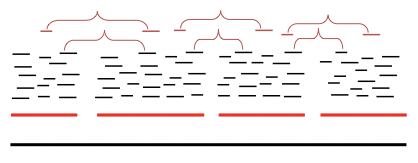
Figura 5.8: Los pares de mate nos ayudan a determinar el orden relativo de los cóntigos para vincularlos en supercóntigos.
A diferencia de los cóntigos, los supercóntigos pueden contener algunos huecos en la secuencia debido a que los pares de relaciones de pareja que conectan los cóntigos solo se secuencian en los extremos. Como generalmente sabemos cuánto tiempo tiene un par de parejas dado, podemos estimar cuántos pares de bases faltan, pero debido a la aleatoriedad de los cortes en la secuenciación de escopeta, es posible que no tengamos los datos disponibles para completar la secuencia exacta. Llenar cada hueco puede ser extremadamente costoso, por lo que incluso los genomas más completamente ensamblados suelen contener algunos huecos.
Derivar secuencia consenso
El objetivo del ensamblaje del genoma es crear una secuencia continua, por lo que después de que las lecturas se hayan alineado en cóntigos, necesitamos resolver cualquier diferencia entre ellas. Como se mencionó anteriormente, algunas de las lecturas superpuestas pueden no ser idénticas debido a errores de secuenciación o polimorfismo. A menudo podemos determinar cuándo ha habido un error de secuenciación cuando una base no está de acuerdo con todas las otras bases alineadas a ella. Teniendo en cuenta los puntajes de calidad en cada una de las bases, normalmente podemos resolver estos conflictos con bastante facilidad. Este método de resolución de conflictos se denomina votación ponderada (Figura 5.9). Otra alternativa es ignorar las frecuencias de cada base y tomar como consenso la letra de máxima calidad. En ocasiones, querrás conservar todas las bases que forman un conjunto polimórfico porque puede ser información importante. En este caso, no podríamos utilizar estos métodos para derivar una secuencia consensuada.
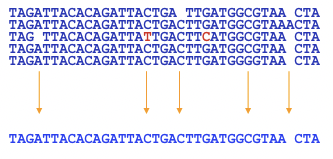
En algunos casos, no es posible derivar un consenso si, por ejemplo, el genoma es heterocigoto y hay números iguales de dos bases diferentes en una ubicación. En este caso, el ensamblador deberá elegir un representante.
¿Sabías?
Dado que el polimorfismo puede complicar significativamente el ensamblaje de genomas diploides, algunos investigadores inducen varias generaciones de endogamia en las especies seleccionadas para reducir la cantidad de heterocigosidad antes de intentar secuenciar el genoma.
En esta sección, vimos un algoritmo para hacer ensamblaje del genoma dadas lecturas. Sin embargo, este algoritmo funciona bien cuando las lecturas tienen una longitud de 500 a 900 bases o más, lo que es típico de la secuenciación de Sanger. Se requieren algoritmos alternos de ensamblaje del genoma es que las lecturas que obtenemos de nuestros métodos de secuenciación son mucho más cortas.