
19.5: La interrupción de los puntos de control del ciclo celular puede causar cáncer
- Page ID
- 54212

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Si un punto de control falla o si una célula sufre daño físico a los cromosomas durante la división celular, o si sufre una mutación somática debilitante en una fase S previa, puede autodestruirse en respuesta a una anomalía bioquímica consecuente. Este es otro ejemplo de apoptosis. Por otro lado, cuando las células mueren por lesión externa, sufren necrosis, una muerte accidental más que programada. En las células que se muestran a continuación, la apoptosis o necrosis fueron inducidas químicamente, seguidas e identificadas como apoptóticas o necróticas usando marcadores fluorescentes (yoduro de propidio, verde; naranja acridina, naranja).
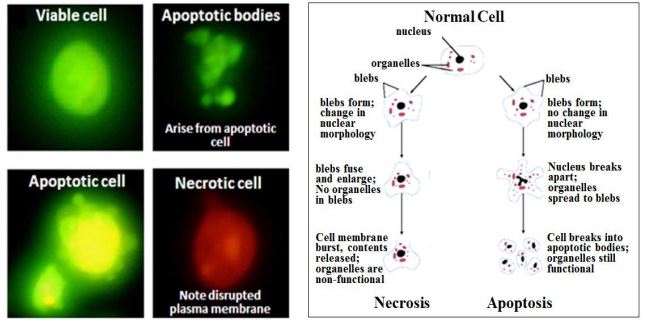
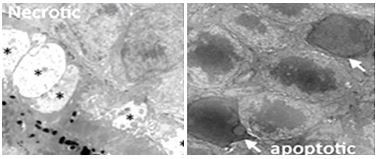
Solo las células con fluorescencia verde (apoptóticas) finalmente formaron cuerpos apoptóticos. En contraste, las células necróticas (naranja-fluorescente) pierden sus membranas plasmáticas, no forman tales 'cuerpos' y eventualmente se desintegrarán. (aumento 400x). Las diferencias en la ultraestructura entre necrosis y apoptosis también se observan en micrografías electrónicas de conos y bastones (izquierda y derecha, respectivamente) a continuación. Un asterisco indica la hinchazón citoplasmática característica del cono necrótico. Las flechas blancas apuntan a núcleos característicos de la apoptosis de células bastonciformes.
Como hemos señalado, las células cíclicas continúan dividiéndose hasta alcanzar G 0 en el estado terminalmente diferenciado. La mayoría de las células terminalmente diferenciadas son eliminadas por apoptosis cuando llegan al final de su vida efectiva, para ser reemplazadas por células madre. También notamos que la señalización accidental puede sacar células de G 0, lo que lleva a una proliferación celular renovada. Si bien estas células son obviamente anormales, no son detectadas por mecanismos de defensa apoptótica. De esta manera, se someten a divisiones celulares incontroladas, convirtiéndose en células cancerosas. Asimismo, las células físicamente dañadas o mutadas a veces pueden escapar del aclaramiento apoptótico. Cuando lo hacen, también pueden convertirse en células cancerosas. El aclaramiento apoptótico y la proliferación incontrolada de células cancerosas se comparan a
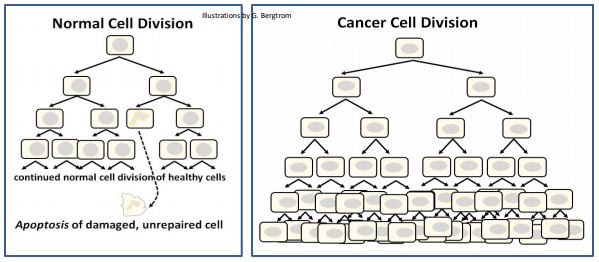
346 Apoptosis (Muerte Celular Programada) vs Necrosis
A. La proteína P53 media el control del ciclo celular normal
El crecimiento canceroso podría resultar si una célula en división normal sufriera una mutación somática que alterara el control normal del ciclo celular. Piense en una sobreexpresión de cdk por ejemplo. Alternativamente, imagine niveles de ciclina en células hijas que nunca bajan; tales células nunca dejarían de ciclar.
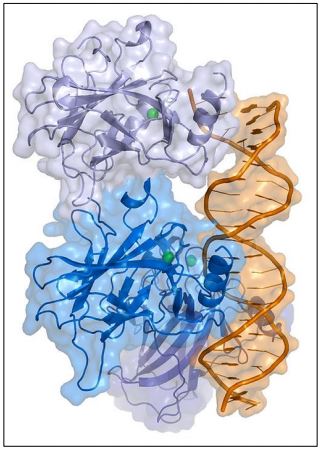
Otras posibilidades incluyen una célula en G 0 que es estimulada para comenzar a ciclar nuevamente por un encuentro inapropiado con una hormona u otra señal. Si no se detectan, estas anomalías pueden transformar las células en células cancerosas. La proteína p53 (ilustrada a continuación) es una proteína reguladora génica de unión al ADN que detecta algunas de estas anomalías y permite que las células en división reparen el daño antes de pasar por los puntos de control del ciclo celular..., o en su defecto, conducirá a la apoptosis de la célula.
No es sorprendente que las mutaciones en el gen para la proteína P53 (llamada TP53 en humanos) estén asociadas con muchos cánceres humanos (pancreático, pulmón, células renales, mama, etc.). Hasta la mitad de los cánceres humanos están asociados con genes p53 mutados. Por lo tanto, p53 es una de una clase de proteínas supresoras de tumores. Los estudios de humanos con una afección conocida como LFS (síndrome de Li-Fraumeni) tienen al menos un alelo p53 mutado. La mutación conduce a un riesgo de cáncer de ~ 100% de por vida, comenzando en la infancia. En las células cultivadas, los genes p53 mutagenizados exhiben características clave de las células cancerosas, incluyendo la proliferación celular no regulada y la supresión de la apoptosis.
1. Cómo funciona p53
La proteína p53 normalmente se une a una proteína Mdm2 activa. Para habilitar los puntos de control del ciclo celular, P53-MDm2 debe separarse y mantenerse separado para permitir que p53 tiempo actúe. En las células en división, el estrés físico o el estrés químico como el daño del ADN durante el crecimiento celular pueden activar una ATM quinasa. ATM quinasa a su vez, fosforila Mdm2, provocando que se disocie de p53. La misma quinasa también fosforila otra quinasa, Chk2, así como la p53 ahora 'libre'. Los eventos iniciados por la ATM cinasa se detallan más adelante.
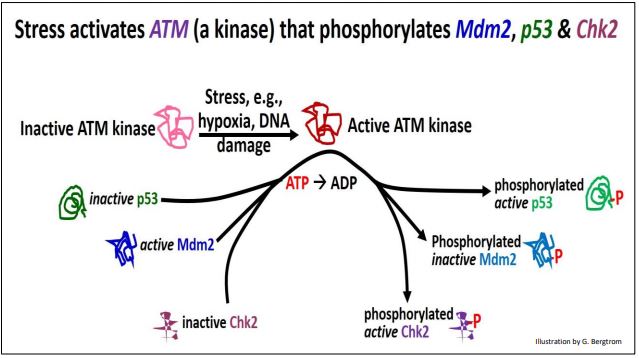
Cada una de las proteínas y enzimas fosforiladas por la ATM quinasa tiene un papel en la función del punto de control del ciclo celular y la detención del ciclo celular mientras se corrigen los errores:
- Ahora separado de Mdm2, fosfo-P53 regula activamente varios genes, incluido el gen p21.
- La proteína P21 se une a cdks; las ciclinas no pueden unirse a las P21-CDK.
- Fosfo-CHK2 activa cataliza la fosforilación de ciclina; las fosfo-ciclinas no pueden unirse a p21-cdks.
- La incapacidad de las ciclinas para unirse a cdks bloquea específicamente el ciclo celular entre las fases G 1 y S, y G 2 a M.
Estos eventos mediados por quinasas en los puntos de control del ciclo celular se ilustran a continuación.
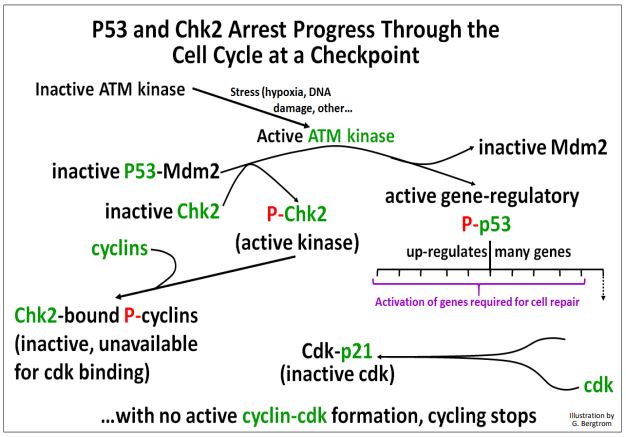
El ciclo celular permanece detenido mientras la célula intenta terminar las actividades bioquímicas esenciales necesarias para corregir aberraciones físicas o químicas inducidas por estrés o cualquier otra aberración antes de pasar a la siguiente fase del ciclo. Si las reparaciones de ADN u otras correcciones son exitosas, la célula puede avanzar a la siguiente fase.
Si no, los proteasomas se dirigen al complejo CHK2-ciclina para la degradación. Asimismo, cualquier P53 que quede unido a Mdm2 no fosforilada también se dirige a la destrucción del proteasoma. El resultado es que cualquier célula incapaz de corregir los efectos del estrés o daño químico, o de reparar el daño del ADN, es blanco para la apoptosis.
Los niveles y actividad de p53 así como las otras proteínas discutidas anteriormente, controlan tanto la cantidad de proteína p53 disponible para responder a anomalías del ciclo celular, como las respuestas mismas. La fosforilación (activación) de p53 no solo conduce a una rápida detención del ciclo celular, sino también a la activación de genes que codifican proteínas requeridas para la reparación del ADN y de proteínas requeridas para la apoptosis (en caso de que falle los esfuerzos de reparación). Las interacciones de p53 con diferentes proteínas que conducen a destinos celulares alternos se resumen a continuación.
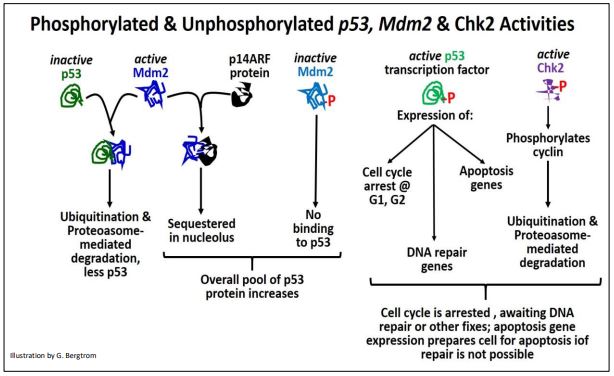
En resumen, p53 suprime el crecimiento de tumores malignos ya sea por
- permitiendo el ADN u otra reparación celular antes de la reanudación del ciclo celular normal, evitando divisiones celulares no reguladas; después de la reparación, p53 y otras proteínas son inactivadas y/o destruidas y el ciclo celular puede reanudarse.
- La incapacidad para reparar/corregir los problemas del ciclo celular pone en movimiento eventos que conducen a la apoptosis, bloqueando así también la tumorigénesis al matar las células dañadas.
Ahora debería quedar claro por qué un mutante p53 que reduzca o elimine la producción de proteína p21 o bloquee la producción esencial de proteínas reparadoras del ADN, permitirá que las células dañadas ingresen a S y las mantengan replicando y dividiendo, transformándolas en células cancerosas. En un giro interesante, parece que en comparación con los humanos, pocas ballenas o elefantes mueren de cáncer, a pesar de tener miles de veces más células que los humanos. La razón parece ser que, al menos para los elefantes, ¡tienen hasta 20 copias (40 alelos) de sus genes p53! Así, una mutación en un alelo de uno de ellos puede tener poco efecto, mientras que prevalecen los efectos reprimentes tumorales de los genes p53 restantes. Lee sobre esta investigación reciente en Whales and Elephants Don't Get Cancer!
2. La centralidad de la acción de p53 en la regulación del ciclo celular
Debido a sus múltiples funciones en la regulación y promoción de la reparación del ADN, y en el control de los puntos de control del ciclo celular, ¡p53 ha sido llamado “el Guardián del Genoma”! Aquí hay más evidencia de este papel central.
a) 'Virus oncogénicos'
Los virus causantes de cáncer incluyen el Virus del Papiloma Humano (VPH), el Virus de Epstein Barr (VEB), el virus de la inmunodeficiencia humana (VIH), los virus de la Hepatitis B y C (VHB, VHC), el virus del herpes humano 8 (HHV-8) y el virus de simios 40 (SV40).
Existe un vínculo demostrado entre SV40, p53 y cáncer. El SV40 es un contaminante viral de las vacunas contra la polio que se utilizaron en la década de 1960. El virus es tumorigénico en mamíferos, aunque no se ha probado una asociación de SV40 y cáncer en humanos. En las células infectadas, el ADN de SV40 ingresa al núcleo donde puede integrarse en el genoma de la célula hospedadora. Las infecciones por SV40 suelen ser latentes, (es decir, no causan ningún daño). Sin embargo, la activación puede conducir a la transformación celular y al crecimiento de sarcomas malignos en músculos así como tumores en otros órganos. La ARN polimerasa II en células infectadas transcribe los genes SV40, produciendo proteínas que replican y encapsulan el ADN viral en una membrana para formar nuevas partículas virales. Sin embargo, el genoma SV40 relativamente pequeño no codifica todas las enzimas y factores necesarios para la replicación del ADN viral. Las propias células infectadas aportan estos factores, produciéndolos solo durante la fase S. En ese momento, el antígeno T grande SV40 (hecho poco después de la infección) ingresa al núcleo de la célula huésped donde regula la transcripción de genes esenciales para la replicación viral y la formación de partículas virales. El antígeno T grande también se une a p53, interfiriendo con la transcripción de proteínas cuyos genes están regulados por p53. Incapaz de ejercer funciones de punto de control, la célula hospedadora se divide incontrolablemente, formando tumores cancerosos. La desregulación del ciclo celular por antígeno T grande asegura el progreso a la fase S y la co-replicación no regulada del ADN viral y de la célula hospedadora.
b) p53 y transducción de señales
El estrés puede activar vías de transducción de señales. Por ejemplo, las mutaciones que afectan a la ruta de señalización de MAPK (MAP quinasa) pueden conducir a la tumorigénesis. Esto puede explicarse por la observación de que cuando se activa, la vía MAPK conduce a la producción amplificada de una quinasa que fosforila p53. La fosfo-p53 activa a su vez aumenta la activación de la vía de transducción de señales MAPK. Tal vez recuerde que la transducción de señales MAPK generalmente termina con una respuesta mitogénica.
Otro ejemplo de interacción p53 es con las proteínas FAK (quinasa de adhesión focal). La actividad de FAK se incrementa por la transducción de señales mediada por integrinas. Recordemos que las integrinas de membrana se unen a la fibronectina, contribuyendo a la formación de la matriz extracelular, o ECM. La elevada actividad FAK participa en la regulación de la adhesión célula-célula y célula-ECM en los puntos de adhesión focales. Otro papel para FAK es unirse directamente a p53 inactiva y aumentar la unión a p53-MDM2. Como acabamos de ver, el P53-MDm2 persistente está dirigido a la ubiquitinación... ¡y la destrucción definitiva! De hecho, los niveles anormalmente altos de FAK están asociados con muchas líneas celulares tumorales diferentes (colon, mama, tiroides, ovario, melanoma, sarcoma...). Estos resultan cuando p53 no puede activar correctamente los puntos de control del ciclo celular.
Si bien las interacciones aquí implícitas son complejas y están bajo estudio activo, estas actividades de p53 ciertamente confirman su papel central como guardián del genoma y como guardián de la división celular.
B. Crecimiento y Comportamiento de las Células Cancer
Los diferentes tipos de células cancerosas tienen diferente crecimiento y otras propiedades conductuales. Es posible que haya oído hablar de cánceres de crecimiento lento y de crecimiento rápido. Los cánceres de colon suelen ser de crecimiento lento. Las colonoscopias periódicas que detectan y extirpan tumores colorrectales en personas de mediana edad o mayores pueden prevenir la enfermedad (aunque los riesgos de enfermedad y el procedimiento en sí deben ser equilibrados). Los cánceres de páncreas son de rápido crecimiento y generalmente pasan desapercibidos hasta llegar a una etapa avanzada. Los objetivos gemelos de la investigación médica son detectar los diferentes cánceres lo suficientemente temprano para una intervención exitosa y, por supuesto, encontrar tratamientos efectivos.
Una sola célula mutada en un tejido puede convertirse en el punto de crecimiento de un tumor, esencialmente una masa de células clonadas a partir de la mutada original. Los tumores o crecimientos benignos (por ejemplo, los fibromas de mama y uterinos en las mujeres, o los lunares comunes en cualquiera de nosotros) dejan de crecer y no ponen en peligro la vida. A menudo se extirpan quirúrgicamente para la comodidad del paciente (o porque las células en algunos tumores benignos pueden tener el potencial de volverse cancerosos).
Los tumores malignos (también llamados neoplasias malignas) son cancerosos y pueden crecer más allá de los límites del propio tumor. Cuando las células tumorales se desprenden pueden ingresar al torrente sanguíneo y viajar a otras partes del cuerpo, el fenómeno llamado metástasis. Las células cancerosas que hacen metástasis pueden convertirse en el punto focal de la formación de nuevos tumores en muchos tejidos diferentes. Debido a que las células cancerosas continúan ciclando y replicando su ADN, pueden sufrir aún más mutaciones somáticas. Estos cambios adicionales pueden facilitar la metástasis y el crecimiento de células cancerosas en diferentes ubicaciones del cuerpo.
C. Estrategias de tratamiento del cáncer
Hay muchos tipos diferentes de cánceres que se originan en diferentes tejidos del cuerpo. Todos ellos comparten la propiedad de la división celular incontrolada, aunque por diferentes razones moleculares y no siempre bien entendidas. Las dos principales estrategias de tratamiento del cáncer desarrolladas en el siglo XX tienen como objetivo interrumpir la replicación de alguna manera.
- La radioterapia se basa en el hecho de que la mayoría de las células de nuestro cuerpo no se dividen, apuntando la radiación mutagénica a los tumores con la esperanza de que el ADN replicante sea mutado en tantos sitios (es decir, genes) que las células tumorales ya no puedan sobrevivir o replicarse adecuadamente.
- La quimioterapia se usa para atacar tumores que no responden bien a la radiación o que no son fácilmente alcanzados por las tecnologías de radiación, y para combatir los cánceres que ni siquiera forman tumores enfocados (como linfomas y leucemias que involucran linfa y células sanguíneas). Estas quimioterapias también tienen como objetivo desviar la replicación o las actividades mitóticas. Por ejemplo, recuerde la cordycepin (didesoxiadenosina trifosfato, o ddATP). Cuando está presente durante la replicación, ddATP se incorpora a una cadena de ADN en crecimiento, después de lo cual no se pueden agregar nucleótidos adicionales a la cadena de ADN. Eso hace que ddATP sea un potente disruptor quimioterapéutico de la replicación. El taxol es otro fármaco de quimioterapia que actúa en este caso, no inhibiendo la replicación de la fase S, sino bloqueando la despolimerización de los microtúbulos de la fibra del huso, bloqueando así la anafase mitótica y la telofase en la última parte de las fases M y C del ciclo. La colchicina (un alcaloide vegetal) ataca las células cancerosas (y otras que se dividen) bloqueando la formación de microtúbulos y, por lo tanto, previniendo la formación de fibra del huso en la profase
Estas terapias no son efectivas contra todos los cánceres y, por supuesto, no se dirigen a tipos específicos de células cancerosas. Su éxito se basa simplemente en el hecho de que las células cancerosas proliferan rápida y constantemente mientras que otros tipos celulares no lo hacen. Muchos, si no todos, los efectos secundarios de la radiación y las quimioterapias son el resultado del daño causado a las células normales en división (por ejemplo, las células del folículo piloso que representan la pérdida de cabello entre muchos pacientes con cáncer, el agotamiento de las células sanguíneas que no pueden ser reemplazadas por células madre en la médula ósea).
Gran parte de la investigación ahora se centra en movilizar el propio sistema inmunológico del cuerpo para crear tratamientos contra el cáncer más específicos y dirigidos. En un poco fascinante de la historia, hace más de 100 años, el Dr. William B. Coley inyectó a un paciente con cáncer terminal una bacteria estreptocócica, que luego emergió libre de tumores al recuperarse de la infección (para más detalles, consulte Los primeros ensayos de inmunoterapia contra el cáncer). Inicialmente se pensó que el fenómeno de “Toxinas del Dr. Coley” era un efecto antitumoral de la bacteria. Pero para 1948 se le atribuyó ampliamente a la respuesta inmune activada por la infección. En la década de 1990, los científicos revisaron la respuesta inmune al cáncer, y para el cambio del siglo XXI, los estudios de inmunoterapia contra el cáncer cobraron fuerza (¡y una financiación de investigación más sustancial!).
Experimentos recientes de inmunoterapia animal y ensayos clínicos en humanos son prometedores. Algunas inmunoterapias ya han sido aprobadas por la FDA de Estados Unidos (Food and Drug Administration). Las estrategias de inmunoterapia contra el cáncer capitalizan el hecho de que su cuerpo a veces reconoce los marcadores de células cancerosas (por ejemplo, moléculas de la superficie celular) como extraños, montando así una defensa inmune contra esas células. Pero esa respuesta a veces no es lo suficientemente poderosa como para despejar nuevas células cancerosas que se dividen rápidamente. El cáncer aparentemente resulta cuando la respuesta inmune es débil. Existen diferentes enfoques, a veces superpuestos, para la inmunoterapia del cáncer. Todos se basan en el hecho de que las células cancerosas que han mutado de alguna manera y están produciendo proteínas aberrantes que el sistema inmunitario puede ver como lo suficientemente extrañas como para provocar una respuesta inmune, por leve que sea. Algunas inmunoterapias buscan potenciar esa respuesta inmune. Otros buscan aislar o generar antígenos celulares cancerosos únicos que inmunizarán a un paciente cuando se les inyecta estos antígenos cancerosos. Algunas inmunoterapias se resumen en la tabla de la página siguiente. Como puede ver en la tabla, las células cancerosas inmuno-dirigidas ya han demostrado ser altamente efectivas. En algunos casos la terapia es un ejemplo de medicina personalizada, en la que los tratamientos se adaptan de manera única a ti como paciente. Los problemas con las inmunoterapias son que
- requieren mucho tiempo y mano de obra, y son costosas de producir.
- si bien pueden 'curarte', es probable que no trabajen en otra persona.
- como la radiación y la quimioterapia, las inmunoterapias vienen con sus propios efectos secundarios desagradables y a veces graves.
Una discusión más detallada sobre las inmunoterapias contra el cáncer se encuentra en el sitio web cancer.gov en Cáncer Tratamiento Inmunotérmico.

NOTA: El término inhibidor de puntos de control en el contexto de las inmunoterapias es diferente al término puntos de control que describen portales para progresar a través del ciclo celular eucariota.