8.6: Direcciones actuales de investigación, ¿qué hemos aprendido? , Bibliografía
- Page ID
- 54695

- Los HMM han sido ampliamente utilizados en diversos campos de la biología computacional. Una de las primeras aplicaciones de este tipo fue en un algoritmo de búsqueda de genes conocido como GENSCAN escrito por Chris Burge y Samuel Karlin [1]. Debido a que la distribución geométrica de la longitud de los HMM no modela bien las regiones exónicas, Burge et al utilizaron una adaptación de HMM conocidos como modelos semi-Markov ocultos (HSMM). Estos tipos de modelos difieren en que cada vez que se alcanza un estado oculto, la duración de ese estado (d i) se elige de una distribución y el estado emite entonces exactamente di caracteres. La transición de este estado oculto al siguiente es entonces análoga al procedimiento HMM excepto que akk = 0 para todos k, evitando así la auto-transición. Muchos de los mismos algoritmos que se desarrollaron previamente para HMM se pueden modificar para HSMM, aunque los detalles no se discutirán aquí, los algoritmos hacia adelante y hacia atrás se pueden modificar para que se ejecuten en el tiempo O (K 2 N 3), donde N es el número de caracteres observados. Esta complejidad temporal supone que no hay límite superior en la duración de un estado, pero imponer tal límite reduce la complejidad a O (K 2 ND 2), donde D es la duración máxima posible de un estado.
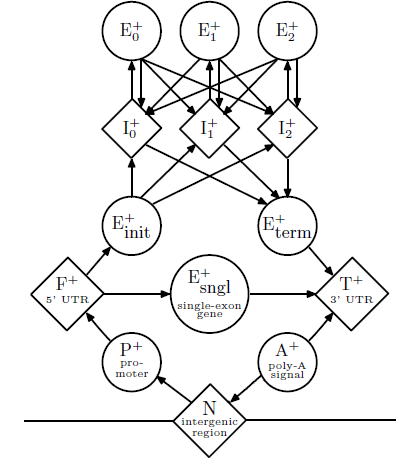
El diagrama de estado básico subyacente al modelo de Burge se representa en la Figura 8.7. El diagrama incluido solo enumera los estados en la cadena directa del ADN, pero en realidad también se incluye una imagen especular de estos estados para la cadena inversa, lo que resulta en un total de 27 estados ocultos. Como ilustra el diagrama, el modelo incorpora muchas de las principales unidades funcionales de genes, incluyendo exones, intrones, promotores, UTR y colas poli-A. Además, se utilizan tres estados intrónicos y exónicos diferentes para asegurar que la longitud total de todos los exones en un gen sea un múltiplo de tres. Similar al ejemplo de la isla CpG, este espacio de estado expandido habilitó la codificación de la memoria dentro del modelo.
- Recientemente se ha hecho un esfuerzo para hacer un enfoque basado en HMM para las búsquedas de homología, llamado HMMER, una alternativa viable a BLAST en términos de eficiencia computacional. A diferencia de la mayoría de los otros algoritmos de búsqueda de homología, HMMER, escrito por Sean Eddy, utiliza la certeza de sobrealineación promedio del algoritmo Forward, en lugar de solo informar la alineación de máxima verosimilitud (a la Viterbi); este enfoque suele ser mejor para detectar homologías más remotas, como tiempos de divergencia aumentar, pueden llegar a ser formas más viables de alinear secuencias, cada una de ellas individualmente no lo suficientemente fuerte como para diferenciarse del ruido sino que juntas dan evidencia de homología. Un desarrollo reciente particularmente emocionante es que HMMER ya está disponible como servidor web; se puede encontrar en http://www.ebi.ac.uk/Tools/hmmer/.
- Un tema interesante que puede explorarse también se refiere a la concordancia de las rutas de decodificación Viterbi y Posterior; no solo para la detección de islas CpG sino incluso para la detección del estado de cromatina. Uno puede mirar múltiples caminos por muestreo, haciendo preguntas como:
— ¿Cuál es el camino máximo a posteriori vs viterbi? ¿Dónde se diferencian?
— ¿Se pueden encontrar rutas completas pero máximamente disjuntas (de Viterbi)?