
23.4: Aplicaciones
- Page ID
- 54614

Análisis de detección de Silico
Con la disponibilidad de una herramienta tan poderosa como FBA, surgen más preguntas de forma natural. Por ejemplo, ¿somos capaces de predecir el fenotipo knockout génico en función de sus efectos simulados sobre el metabolismo? Además, ¿por qué trataríamos de hacer esto, a pesar de que existen otros métodos, como el mapa de interacción de proteínas conectivo? Dicho análisis es realmente necesario, ya que otros métodos no toman en consideración directa el flujo metabólico u otras condiciones metabólicas específicas.
Licencia Commons. Para obtener más información, consulte http://ocw.mit.edu/help/faq-fair-use/.
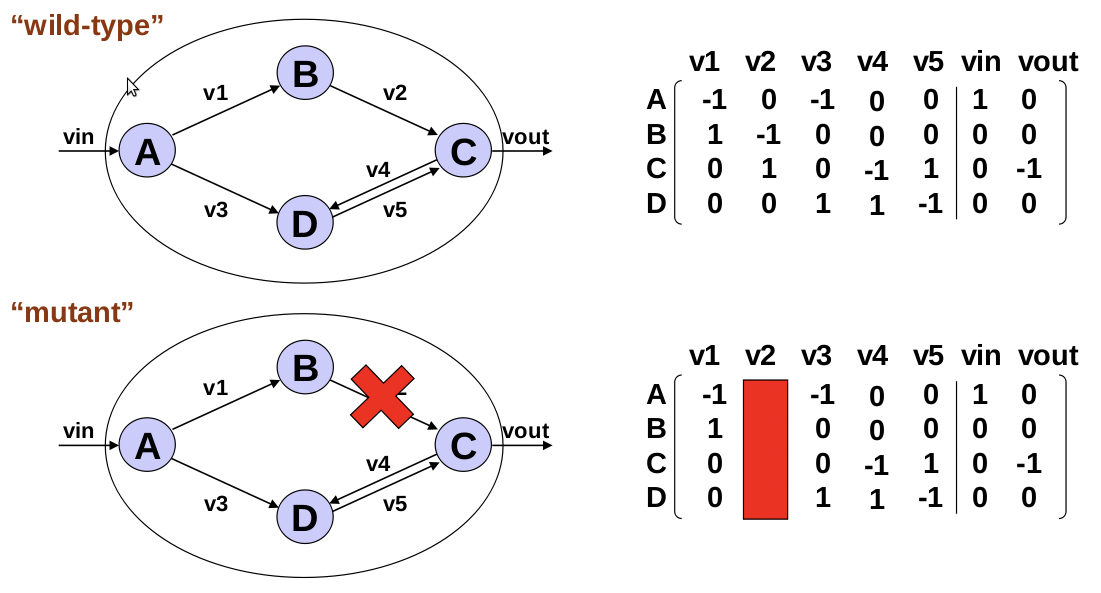
Figura 23.4: Eliminar una reacción es lo mismo que eliminar un gen de la matriz estequiométrica.
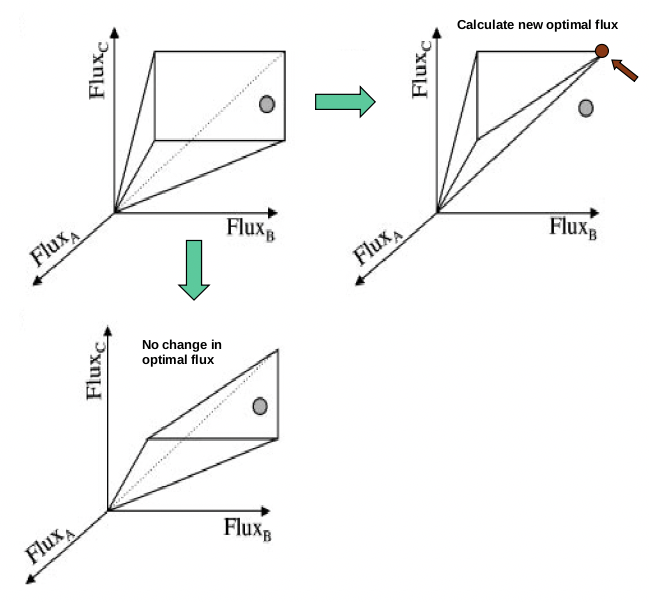
La noqueación de un gen en un experimento se modela simplemente eliminando una de las columnas (reacciones) de la matriz estequiométrica. (Una pregunta durante la clase aclaró que un solo gen puede noquear múltiples columnas/reacciones). De este modo, estas mutaciones knockout limitarán aún más el espacio de solución factible al eliminar los flujos y sus vías extremas relacionadas. Si el flujo óptimo original estaba afuera está fuera del nuevo espacio, entonces se crea un nuevo flujo óptimo. Así, el análisis de FBA producirá diferentes soluciones. La solución es una tasa de crecimiento máxima, la cual puede ser confirmada o desprobada experimentalmente. La tasa de crecimiento en la nueva solución proporciona una medida del fenotipo knockout. Si estos knockouts de genes son de hecho letales, entonces la solución óptima será una tasa de crecimiento de cero.
Estudios de Edwards, Palsson (1900) exploran el uso de la predicción de fenotipos knockout para predecir cambios metabólicos en respuesta a la eliminación de enzimas en E. coli, un procariota [? ]. En otras palabras, se construyó un modelo metabólico in silico de E. coli para simular mutaciones que afectan las vías de glucólisis, fosfato de pentosa, TCA y transporte de electrones (436 metabolitos y 719 reacciones incluidas). Para cada condición específica, se comparó el crecimiento óptimo de mutantes con los no mutantes. Luego se compararon los resultados in vivo e in silico, con 86% de acuerdo. Los errores en el modelo indican un modelo subdesarrollado (falta de conocimiento). Los autores discuten 7 errores no modelados por FBA, incluyendo mutantes que inhiben la síntesis estable de ARN y producen intermedios tóxicos.
Licencia Commons. Para obtener más información, consulte http://ocw.mit.edu/help/faq-fair-use/.
Figura 23.5: La restricción del espacio de solución factible puede crear un nuevo flujo óptimo.
Predicciones del modelo de flujo cuantitativo en si
¿Pueden los modelos predecir cuantitativamente los flujos, la tasa de crecimiento? Demostramos la capacidad de FBA para dar predicciones cuantitativas sobre la tasa de crecimiento y los flujos de reacción dadas las diferentes condiciones ambientales. Más específicamente, la predicción se refiere a flujos medibles externamente en función de las tasas de absorción controladas y las condiciones ambientales. Dado que FBA maximiza una función objetiva, dando como resultado un valor específico para esta función, en teoría deberíamos poder extraer información cuantitativa del modelo.
Un ejemplo temprano de Edwards, Ibarra y Palsson (1901), predijo la tasa de crecimiento de E. coli en cultivo dado un rango de tasas fijas de absorción de oxígeno y dos fuentes de carbono (acetato y succinato), que podrían controlar en un reactor discontinuo [6]. Supusieron que las células de E. coli ajustan su metabolismo para maximizar el crecimiento (usando una función objetivo de crecimiento) bajo condiciones ambientales dadas y utilizaron FBA para modelar las vías metabólicas en la bacteria. La entrada a este modelo en particular es el acetato y el oxígeno, el cual está etiquetado como VIN.
Las tasas de absorción controladas fijaron los valores de los flujos de entrada de oxígeno y acetato/succinato en la red, pero los otros flujos se calcularon para maximizar el valor del objetivo de crecimiento.
La tasa de crecimiento aún se trata como la solución al análisis de FBA. En suma, la tasa de crecimiento óptima se predice en función de las restricciones de absorción de oxígeno versus acetato y oxígeno versus succinato. El modelo básico es una línea predictiva y puede confirmarse experimentalmente en un biorreactor midiendo la captación y el crecimiento de reactores discontinuos (nota: la captación experimental no fue restringida, solo se midió).
Este modelo de Palsson fue la primera buena prueba de principio in silico modelo. Las predicciones cuantitativas de la tasa de crecimiento de los autores bajo las condiciones dierentes coincidieron muy estrechamente con las tasas de crecimiento observadas experimentalmente, lo que implica que E. coli sí tiene una red metabólica que está diseñada para maximizar el crecimiento. Tuvo buenas tasas verdaderas positivas y verdaderas negativas. La concordancia entre las predicciones y los resultados experimentales es muy impresionante para un modelo que no incluye ninguna información cinética, solo estequiometría. El profesor Galagan advirtió, sin embargo, que a menudo es difícil saber qué es el buen acuerdo, porque desconocemos la importancia del tamaño de los residuos. El organismo se cultivó sobre una serie de nutrientes dierentes. Por lo tanto, los investigadores pudieron predecir el crecimiento específico de la condición. Ten en cuenta que esto funcionó, ya que solo ciertos genes son necesarios para algunos nutrientes, como fbp para la gluconeogénesis. Por lo tanto, noquear fbp solo será letal cuando no haya glucosa en el ambiente, condición específica que resultó en una solución de crecimiento al ser analizada por FBA.
Modelado de estado cuasi estacionario (QSSM)
Ahora podemos describir cómo usar FBA para predecir cambios dependientes del tiempo en las tasas de crecimiento y las concentraciones de metabolitos usando modelos cuasi de estado estacionario. En el ejemplo anterior se utilizó FBA para hacer predicciones cuantitativas de crecimiento bajo condiciones ambientales específicas (predicciones puntuales). Ahora, después de los flujos de crecimiento y captación, pasamos a otra suposición y tipo de modelo.
¿Podemos usar un modelo de metabolismo en estado estacionario para predecir los cambios dependientes del tiempo en la célula o los entornos? Tenemos que hacer una serie de supuestos cuasi de estado estacionario (QSSA):
- El metabolismo se ajusta a los cambios ambientales/celulares más rápidamente que los cambios en sí
- Las concentraciones celulares y ambientales son dinámicas, pero el metabolismo opera con la condición de que la concentración sea estática en cada punto temporal (modelo de estado estacionario).
¿Es posible utilizar QSSM para predecir la dinámica metabólica a lo largo del tiempo? Por ejemplo, si se toma menos acetato por célula a medida que crece el cultivo, entonces la tasa de crecimiento debe disminuir. Pero ahora, se aplican los supuestos de QSSA. Es decir, en efecto, en cualquier momento dado, el organismo se encuentra en estado estacionario.
¿Qué valores obtiene uno como solución al problema de FBA? Hay flujos de la tasa de crecimiento. Estamos predicando tasa y flujos (solución) donde VIN/OUT incluyó. Hasta ahora suponíamos que la entrada y salida son infinitos sumideros y fuentes. Para modelar la dinámica de sustrato/crecimiento, el análisis se realiza de manera un poco diferente al análisis de flujo cuantitativo previo. Primero dividimos el tiempo en cortes t. En cada punto de tiempo t, usamos FBA para predecir la captación de sustrato celular (Su) y el crecimiento (g) durante el intervalo t. El QSSA significa que estas predicciones son constantes sobre t. Luego, integramos para obtener la biomasa (B) y la concentración de sustrato (Sc) en el siguiente punto de tiempo t + t. Por lo tanto, el nuevo VIN se calcula cada vez en función de los puntos t de tiempo intermedio. Así podemos predecir la tasa de crecimiento y la captación de glucosa y acetato (nutrientes disponibles en el ambiente). El análisis de cuatro pasos es:
- La concentración en el tiempo viene dada por la concentración de sustrato de la última etapa más cualquier sustrato adicional proporcionado al cultivo celular por un flujo de entrada, tal como en un lote alimentado.
- La concentración de sustrato se escala por tiempo y biomasa (X) para determinar la disponibilidad de sustrato para las células. Esto puede exceder la tasa máxima de captación de las células o ser menor que ese número.
- Utilice el modelo de balance de flujo para evaluar la tasa de absorción real del sustrato, que puede ser mayor o menor que el sustrato disponible según lo determinado por la etapa 2.
- La concentración para el siguiente paso de tiempo se calcula integrando las ecuaciones diferenciales estándar:
\ [\ frac {d B} {d t} =g B\ fila derecha B=B_ {o} e^ {g t}\ nonúmero]
\[\frac{d S c}{d t}=-S u B \rightarrow S c=S c_{o} \frac{X}{g}\left(e^{g t}-1\right)\nonumber\]
El trabajo adicional de Varma et al. (1994) especifica la tasa de captación de glucosa a priori [17]. Las simulaciones del modelo funcionan para predecir cambios dependientes del tiempo en el crecimiento, la absorción de oxígeno y la secreción de acetato. Este modelo inverso traza las tasas de absorción versus el crecimiento, mientras que aún logra resultados comparables in vivo e in silico. Los investigadores utilizaron modelos cuasi de estado estacionario para predecir los perfiles dependientes del tiempo de crecimiento celular y concentraciones de metabolitos en cultivos discontinuos de E. coli que tenían un suministro inicial limitado de glucosa (izquierda) o un suministro continuo lento de glucosa (diagrama derecho). Un gran ajuste es evidente.
Los diagramas anteriores muestran los resultados de las predicciones del modelo (líneas continuas) y lo comparan con los resultados experimentales (puntos individuales). Así, en E. coli, las predicciones cuasiestables son impresionantemente precisas incluso con un modelo que no tiene en cuenta ningún cambio en los niveles de expresión enzimática a lo largo del tiempo. Sin embargo, este modelo no sería adecuado para describir comportamientos que se sabe que implican regulación génica. Por ejemplo, si las células hubieran sido cultivadas en medio de media glucosa/mitad lactosa, el modelo no habría podido predecir el cambio en el consumo de una fuente de carbono a otra. (Esto ocurre experimentalmente cuando E. coli activa vías alternas de utilización de carbono solo en ausencia de glucosa).
Regulación vía lógica booleana
Hay una serie de niveles de regulación a través de los cuales se controla el flujo metabólico en los niveles metabolitos, transcripcionales, traduccionales, postraduccionales. Los errores asociados a FBA pueden explicarse por la incorporación de información reguladora de genes en los modelos. Una forma de hacerlo es la lógica booleana. La siguiente tabla describe si los genes para enzimas asociadas están activados o apagados en presencia de ciertos nutrientes (un ejemplo de incorporación de las preferencias de E. coli mencionadas anteriormente):
| ON sin glucosa (0) |
EN acetato presente (1) |
|
EN glucosa presente (1) |
OFF acetato presente (1) |
Por lo tanto, se puede pensar que el siguiente paso a dar es incorporar este hecho a los modelos. Por ejemplo, si tenemos glucosa en el ambiente, los genes relacionados con el procesamiento de acetato están apagados y por lo tanto ausentes de la matriz S que ahora se vuelve dinámica como resultado de la incorporación de la regulación en nuestro modelo. Al final, nuestro modelo no es cuantitativo. La regulación básica describe entonces que si una enzima procesadora de nutrientes está encendida, la otra está apagada. Básicamente se trata de un montón de lógica booleana, basada en la presencia de enzimas, metabolitos, genes, etc. Estas suposiciones de estilo booleano se utilizan luego en cada pequeño cambio en el tiempo dt para evaluar la tasa de crecimiento, los flujos y tales variables. Entonces, dados los flujos predichos, el VIN, el VOUT y los estados del sistema, se puede usar la lógica para apagar y encender genes, efectivamente una S por t. Podemos comenzar a armar todos los análisis anteriores y llegar a un enfoque general en el modelado metabólico. Podemos decir que si la glucólisis está encendida, entonces la gluconeogénesis debe estar apagada.
El primer intento de incluir la regulación en un modelo de FBA fue publicado por Covert, Schilling y Palsson en 1901 [7]. Los investigadores incorporaron un conjunto de eventos reguladores transcripcionales conocidos en su análisis de una red reguladora metabólica al aproximar la regulación génica como un proceso booleano. Se dijo que se producía o no una reacción dependiendo de la presencia tanto de la enzima como del sustrato (s): si la enzima que cataliza la reacción (E) no se expresa o un sustrato (A) no está disponible, el flujo de reacción será cero:
rxn = SI (A) Y (E)
La lógica booleana similar determinó si las enzimas se expresaban o no, dependiendo de los genes expresionados actualmente y las condiciones ambientales actuales. Por ejemplo, la transcripción de la enzima (E) ocurre solo si el gen apropiado (G) está disponible para la transcripción y no está presente ningún represor (B):
trans = SI (G) Y NO (B)
Los autores utilizaron estos principios para diseñar una red booleana que introduce el estado actual de todos los genes relevantes (encendido o apagado) y el estado actual de todos los metabolitos (presentes o no presentes), y emite un vector binario que contiene el nuevo estado de cada uno de estos genes y metabolitos. Las reglas de la red booleana se construyeron a partir de eventos regulatorios celulares determinados experimentalmente. El tratamiento de las reacciones y las concentraciones de enzimas/metabolitos como variables binarias no permite el análisis cuantitativo, pero este método puede predecir cambios cualitativos en los flujos metabólicos cuando se fusionan con FBA. Siempre que una enzima está ausente, la columna correspondiente se retira de la matriz de reacción de FBA, como se describió anteriormente para la predicción del fenotipo knockout. Esto lleva a un proceso iterativo:
1. Dados los estados iniciales de todos los genes y metabolitos, calcular los nuevos estados usando la red booleana;
2. realizar FBA con columnas apropiadas eliminadas de la matriz, con base en los estados de las enzimas, para determinar las nuevas concentraciones de metabolitos;
3. repetir el cálculo de la red booleana con las nuevas concentraciones de metabolitos; etc. El modelo anterior no es cuantitativo, sino más bien una simulación pura de activar y desactivar genes en cualquier momento en particular instante.
En algunas reacciones metabólicas, existen reglas sobre permitir que el organismo cambie las fuentes de carbono (C1, C2).
Una aplicación de este método del estudio de Covert et al. [7] fue simular el desplazamiento diáuxico, un cambio de metabolizar una fuente de carbono preferida a otra fuente de carbono cuando la fuente preferida no está disponible. El proceso modelado incluye dos productos génicos, una proteína reguladora RPc1, que detecta (es activada por) el Carbono 1, y una proteína de transporte Tc2, que transporta el Carbono 2. Si RpC1 es activado por el Carbono 1, Tc2 no se transcribirá, ya que la célula utiliza preferentemente el Carbono 1 como fuente de carbono. Si el Carbono 1 no está disponible, la célula cambiará a vías metabólicas basadas en Carbono 2 y activará la expresión de Tc2.
Los booleanos pueden representar esta información:
RpC1 = IF (Carbon1) Tc2 = SI NO (RPC1)
Covert et al. encontraron que este enfoque dio predicciones sobre el metabolismo que coincidieron con los resultados del cambio diáuxico inducido experimentalmente. Este desplazamiento diáuxico está bien modelado por el análisis in silico ver figura anterior. En el segmento A, C1 se agota como nutriente y hay crecimiento. En el segmento B, no hay crecimiento ya que C1 se ha agotado y aún no se elaboran las enzimas procesadoras C2, ya que los genes no se han activado (o están en proceso), de ahí el retraso de la cantidad constante de biomasa. En el segmento C, las enzimas para C2 se encendieron y la biomasa aumenta a medida que el crecimiento continúa con una nueva fuente de nutrientes. Por lo tanto, si no hay C1, C2 se agota. A medida que C1 se agota, el organismo desplaza la actividad metabólica a través de la regulación genética y comienza a tomar C2. Regulación predice diauxie, el uso de C1 antes de C2. Sin regulación, el sistema crecería tanto en C1 como en C2 juntos para obtener un máximo de biomasa.
Hasta el momento hemos discutido el uso de este enfoque combinado de la red FBA-Booleana para modelar la regulación a nivel transcripcional/traduccional, y también funcionará para otros tipos de regulación. La principal limitación es para formas lentas de regulación, ya que este método supone que los pasos regulatorios se completan dentro de un solo intervalo de tiempo (porque el cálculo booleano se realiza en cada paso de tiempo de FBA y no toma en cuenta estados previos del sistema). Esto está bien para cualquier forma de regulación que actúe al menos tan rápido como la transcripción/traducción. Por ejemplo, la fosforilación de enzimas (un proceso de activación enzimática) es muy rápida y puede modelarse incluyendo la presencia de una enzima fosforilasa en la red booleana.
Sin embargo, la regulación que ocurre en escalas de tiempo más largas, como el secuestro de ARNm, no es tomada en cuenta por este modelo. Este enfoque también tiene un problema fundamental en el sentido de que no permite introducir mediciones experimentales reales de los niveles de expresión génica en momentos relevantes.
No necesitamos nuestras simulaciones para predecir artificialmente si ciertos genes están activados o apagados. Los datos de expresión de microarrays nos permiten determinar qué genes se están expresando, y esta información se puede incorporar a nuestros modelos.
Acoplamiento de la expresión génica con el metabolismo
En la práctica, no necesitamos modelar artificialmente los niveles de genes, podemos medirlos. Como se discutió anteriormente, es posible medir los niveles de expresión de todos los ARNm en una muestra dada. Dado que los datos de expresión de ARNm se correlacionan con los datos de expresión de proteínas, sería extremadamente útil incorporarlos en el FBA. Por lo general, los datos de experimentos de microarrays se agrupan, y se plantea la hipótesis de que los genes desconocidos tienen una función similar a la función de aquellos genes conocidos con los que se agrupan. Este análisis puede ser defectuoso, sin embargo, ya que los genes con acciones similares pueden no siempre agruparse. La incorporación de datos de expresión de microarrays en FBA podría permitir un método alternativo de interpretación de los datos. Aquí surge una pregunta, ¿cuál es la relación entre el nivel génico y el flujo a través de una reacción?
Di la reacción ¡A! B es catalizado por una enzima. Si hay mucha A presente, el aumento de la expresión del gen para la enzima provoca un aumento de la velocidad de reacción. De lo contrario, aumentar el nivel de expresión génica no aumentará la velocidad de reacción. Sin embargo, la concentración enzimática puede tratarse como una restricción sobre el flujo máximo posible, dado que el sustrato también tiene un límite fisiológico razonable.
El siguiente paso, entonces, es relacionar el nivel de expresión de ARNm con la concentración de enzima. Esto es más difícil, ya que las células tienen una serie de mecanismos reguladores para controlar las concentraciones de proteínas independientemente de las concentraciones de ARNm. Por ejemplo, las proteínas traducidas pueden requerir una etapa de activación adicional (por ejemplo, fosforilación), cada molécula de ARNm puede traducirse en un número variable de proteínas antes de que se degrada (por ejemplo, mediante ARN antisentido), la tasa de traducción del ARNm a la proteína puede ser más lenta que los intervalos de tiempo considerados. en cada etapa de FBA, y la tasa de degradación de proteínas también puede ser lenta. A pesar de estas complicaciones, los niveles de expresión de ARNm de los experimentos de micromatrices generalmente se toman como límites superiores en las posibles concentraciones de enzimas en cada punto de tiempo medido. Dada la relación anterior entre la concentración enzimática y el flujo, esto significa que los niveles de expresión de ARNm también son límites superiores en los flujos máximos posibles a través de las reacciones catalizadas por sus proteínas codificadas. La validez de este supuesto aún se está debatiendo, pero ya se ha desempeñado bien en los análisis de FBA y es consistente con evidencia reciente de que las células sí controlan los niveles de enzimas metabólicas principalmente ajustando los niveles de ARNm. (En 1907, el profesor Galagan discutió un estudio de Zaslaver et al. (1904) que encontró que los genes requeridos en una ruta de biosíntesis de aminoácidos se transcriben secuencialmente según sea necesario [2]). Esta es una suposición particularmente útil para incluir datos de expresión de micromatrices en FBA, ya que FBA hace uso de valores de flujo máximo para limitar el cono de equilibrio de flujo.
Colijn et al. abordan la cuestión de la integración algorítmica de datos de expresión y redes metabólicas [3]. Aplican FBA para modelar el flujo máximo a través de cada reacción en una red metabólica. Por ejemplo, si se dispone de datos de micromatrices de un organismo que crece en glucosa y de un organismo que crece en acetato, es probable que se observen diferencias reguladoras significativas entre los dos conjuntos de datos. Vmax nos dice cuál es el máximo que podemos alcanzar. Microarray detecta el nivel de transcripciones, y da un límite superior de Vmax.
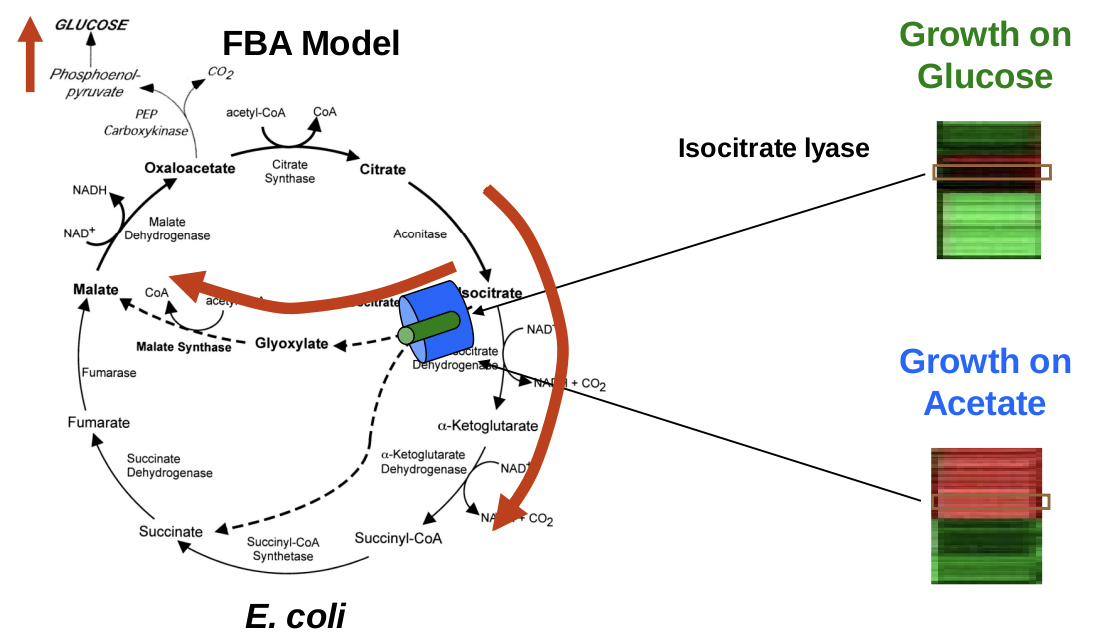
Además de predecir vías metabólicas bajo diferentes condiciones ambientales, los experimentos de FBA y microarrays se pueden combinar para predecir el estado de un sistema metabólico bajo diferentes tratamientos farmacológicos. Por ejemplo, varios medicamentos contra la tuberculosis se dirigen a la biosíntesis de ácido micólico. El ácido micólico es un constituyente principal de la pared celular. En un artículo de 1904 de Boshode et al., los investigadores probaron 75 fármacos, combinaciones de fármacos y condiciones de crecimiento para
Cortesía de los autores. Licencia: CC BY.
Fuente: Colijin, Caroline, et al. “Interpretación de datos de expresión con modelos de flujo metabólico: predicción de Mycobacterium
Tubercluosis Producción de ácido micólico.” PLoS Biología Computacional 5, núm. 8 (2009): e1000489
ver qué efecto tuvieron los diferentes tratamientos sobre la síntesis de ácido micólico [9]. En 1905, Raman et al. publicaron un modelo FBA de biosíntesis de ácido micólico, consistente en 197 metabolitos y 219 reacciones [13].
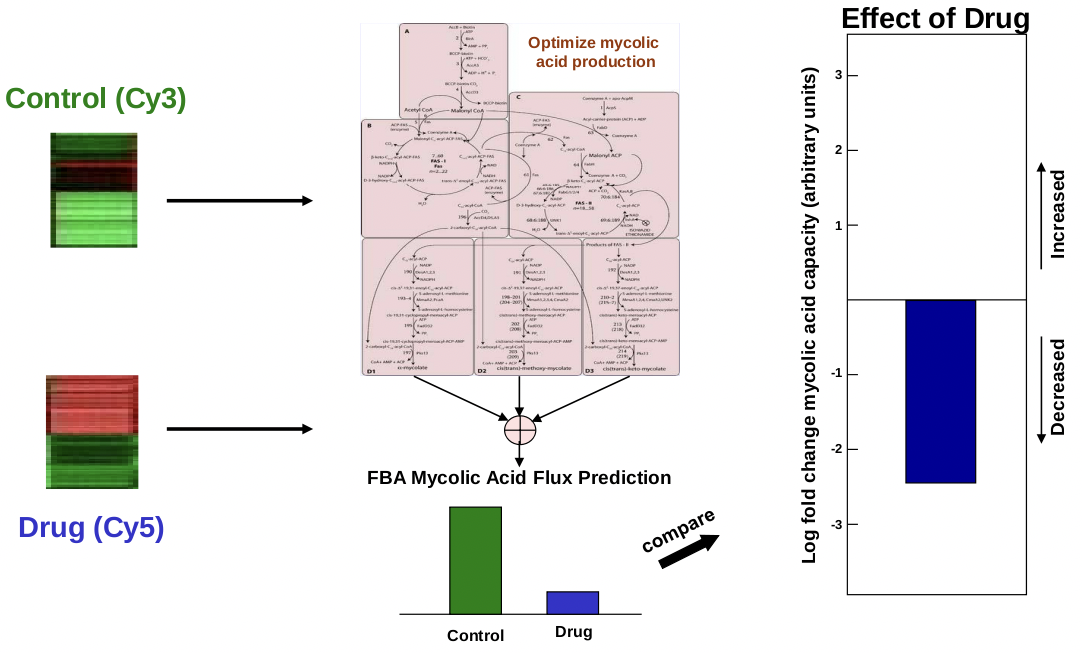
El flujo básico de la predicción fue tomar un valor de expresión control y un valor de expresión de tratamiento para un conjunto particular de genes, luego alimentar esta información al FBA y medir el efecto final sobre el tratamiento sobre la producción de ácido micólico. Para examinar los inhibidores y potenciadores predichos, examinaron la significación, que examina si el efecto se debe al ruido, y la especificidad, que examina si el efecto se debe al ácido micólico o a la supresión/mejora global del metabolismo. Los resultados fueron bastante alentadores. Varios inhibidores conocidos del ácido micólico fueron identificados por el FBA. También se encontraron resultados interesantes entre fármacos no conocidos específicamente para inhibir la síntesis de ácido micólico. Se predijeron 4 nuevos inhibidores y 2 nuevos potenciadores de la síntesis de ácido micólico. Un fármaco en particular, Triclosán, parece ser un potenciador según el modelo FBA, mientras que actualmente se le conoce como inhibidor. Sería interesante estudiar más a fondo esta droga en particular. Las pruebas experimentales y la validación están actualmente en curso.
La agrupación también puede ser ineficaz para identificar la función de diversos tratamientos. Los inhibidores predichos y los potenciadores predichos de la síntesis de ácido micólico no se agrupan entre sí. Además, no se requiere un conjunto de entrenamiento etiquetado para la clasificación algorítmica basada en la FBA, mientras que es necesario para los algoritmos de agrupamiento supervisados.
Predicción de la fuente de nutrientes
Ahora, tenemos la idea de predecir la fuente de nutrientes que un organismo puede estar usando en un ambiente, observando los datos de expresión y buscando la expresión génica asociada al procesamiento de nutrientes. Esto es más fácil, ya que no podemos entrar al medio ambiente y medir todos los niveles químicos, pero podemos obtener datos de expresión con bastante facilidad. Es decir, tratamos de predecir una fuente de nutrientes a través de predicciones del estado metabólico a partir de datos de expresión, con base en el supuesto de que es probable que los organismos ajusten el estado metabólico a los nutrientes disponibles. Luego, los nutrientes pueden clasificarse según qué tan bien coinciden con los estados metabólicos.
Al revés también podría funcionar. ¿Puedo predecir un nutriente dado un estado? Tales predicciones podrían ser útiles para determinar los requerimientos nutrimentales de un organismo con un entorno natural desconocido, o para determinar cómo un organismo cambia su ambiente. (La TB, por ejemplo, es capaz de vivir dentro del ambiente de un fagolisosoma macrófago, presumiblemente alterando las condiciones ambientales en el
Cortesía de los autores. Licencia: CC BY.
Fuente: Coljin, Caroline, et al. “Interpretación de datos de expresión con modelos de flujo metabólico: predicción
Producción de ácido micólico por Mycobacterium Tuberculosis.” PLoS Biología Computacional 5, núm. 8 (2009): e1000489
Podemos usar FBA para definir un espacio de posibles estados metabólicos y elegir uno. Los pasos básicos son:
• Comience con el cono de flujo máximo (representando el mejor crecimiento con todos los nutrientes disponibles en el ambiente). Encuentre un flujo óptimo para cada nutriente.
• Aplicar conjunto de datos de expresión (aún sin conocer nutriente). Esto le permitirá limitar la forma del cono y averiguar el nutriente, que se representa como uno con la distancia más cercana a la solución óptima.
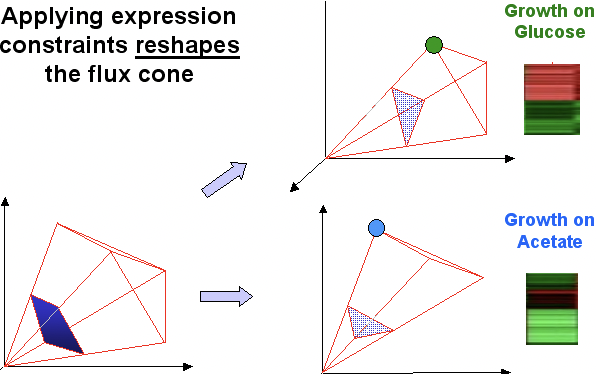
Licencia Commons. Para obtener más información, consulte http://ocw.mit.edu/help/faq-fair-use/.
Figura 23.8: La aplicación del conjunto de datos de expresión permite restringir la forma del cono.
En la Figura 8, puede ver que el primer cono tiene una serie de óptimos, por lo que se desconoce el nutriente real. Sin embargo, después de aplicar los datos de expresión, el cono es remodelado. Tiene solo un óptimo, el cual se encuentra aún en un espacio factible y con ello debe ser ese nutriente que estás buscando.
Como antes, los niveles de expresión medidos proporcionan restricciones en los flujos de reacción, alterando la forma del cono de equilibrio de flujo (ahora el cono de equilibrio de flujo restringido por expresión). FBA se puede usar para determinar el conjunto óptimo de flujos que maximizan el crecimiento dentro de estas restricciones de expresión, y este conjunto de flujos se puede comparar con patrones de crecimiento óptimos determinados experimentalmente bajo cada condición ambiental de interés. La diferencia entre el estado calculado del organismo y el estado óptimo bajo cada condición es una medida de cuán subóptimo sería el estado metabólico actual del organismo si de hecho estuviera creciendo bajo esa condición.
Licencia Commons. Para obtener más información, consulte http://ocw.mit.edu/help/faq-fair-use/.
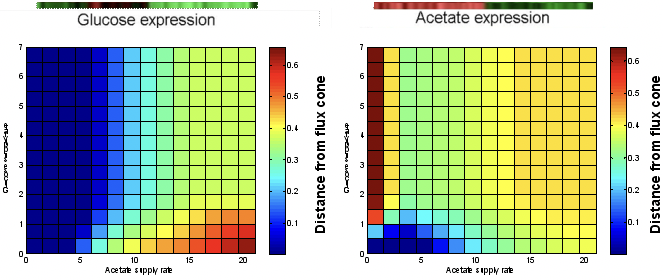
Figura 23.9: Resultados del experimento de predicción de fuentes nutrimentales.
Los datos de expresión del crecimiento y el metabolismo se pueden aplicar para predecir la fuente de carbono que se está utilizando. Por ejemplo, considere el producto nutritivo de E. coli. Podemos simular este sistema para glucosa versus acetato. El color indica la distancia desde el cono de flujo restringido hasta la solución de flujo óptimo para ese combo de nutrientes (mismo procedimiento descrito anteriormente). Entonces, se pueden clasificar múltiples nutrientes, priorizados de acuerdo con los datos de expresión. Datos inéditos de Desmond Lun y Aaron Brandes proporcionan un ejemplo de este enfoque.
Utilizaron FBA para predecir en qué fuente nutritiva estaban creciendo los cultivos de E. coli, con base en datos de expresión génica. Compararon los flujos óptimos conocidos (el punto óptimo en el espacio de flujo) para cada condición nutritiva con los valores de flujo óptimos permitidos dentro del cono de equilibrio de flujo restringido por expresión. Aquellas condiciones nutritivas con flujos óptimos que permanecieron dentro (o más cercanos a) el cono de expresión restringida fueron las posibilidades más probables para el ambiente real del cultivo.
Los resultados del experimento se muestran en la Figura 9, donde cada cuadrado en las matrices de resultados se colorea con base en la distancia entre los flujos óptimos para esa condición nutritiva y los flujos óptimos calculados con base en los datos de expresión. Los valores rojos indican grandes distancias desde el cono de flujo con restricción de expresión y los valores azules indican distancias cortas desde el cono. En los experimentos de glucosa-acetato, por ejemplo, los resultados del experimento de la izquierda indican que las condiciones bajas de acetato son las más probables (y la glucosa fue el nutriente en el cultivo) y los resultados del experimento de la derecha indican que las condiciones bajas en glucosa/acetato medio son las más probables ( y el acetato fue el nutriente en el cultivo). Cuando se consideraron 6 nutrientes posibles, el modelo siempre predijo el correcto, y cuando se consideraron 18 nutrientes posibles, el correcto siempre fue una de las 4 mejores predicciones de clasificación. Estos resultados sugieren que es posible utilizar datos de expresión y modelos FBA para predecir condiciones ambientales a partir de información sobre el estado metabólico de un organismo.
Esto es importante porque la TB utiliza ácidos grasos en macrófagos en los sistemas inmunitarios. No sabemos cuáles son exactamente los que se utilizan. Podemos averiguar lo que la TB ve en su entorno como fuente de alimento y factor de proliferación analizando qué genes de procesamiento de nutrientes relacionados se encienden en las fases de crecimiento y similares. De esta manera podemos averiguar los nutrientes que necesita para crecer, permitiendo una forma potencial de matarlo al no suministrarle dichos nutrientes o noqueando esos genes en particular.
Es más fácil obtener datos de expresión para ver la actividad del flujo que ver qué se está agotando en el ambiente analizando la química en un nivel tan pequeño. Además, es posible que no podamos cultivar algunas bacterias en el laboratorio, pero podemos resolver el problema obteniendo los datos de expresión de las bacterias que crecen en un ambiente natural y luego ver qué está usando para crecer. Entonces, podemos agregarlo al medio de laboratorio para cultivar las bacterias con éxito.