
3.8: Lisosomas y Peroxisomas
- Page ID
- 56612

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Una célula está compuesta por muchos orgánulos diferentes y microcuerpos (o citosomas) es un tipo de orgánulo que se encuentra en las células de plantas, protozoos y animales. Los orgánulos de la familia de los microcuerpos incluyen peroxisomas, glioxisomas, glucosomas e hidrogenosomas.
Lisosomas
Los lisosomas son cuerpos aproximadamente esféricos encerrados por una sola membrana. Son fabricados por el aparato de Golgi (Figura\(\PageIndex{1}\)) y contienen más de 50 tipos diferentes de enzimas hidrolíticas incluyendo proteasas, lipasas, nucleasas y polisacaridasas. El pH dentro del lisosoma es aproximadamente pH 5, sustancialmente menor que el del citosol (~pH 7.2). Todas las enzimas en el lisosoma funcionan mejor a un pH ácido, lo que reduce el riesgo de que digieran su propia célula si deben escapar del lisosoma.
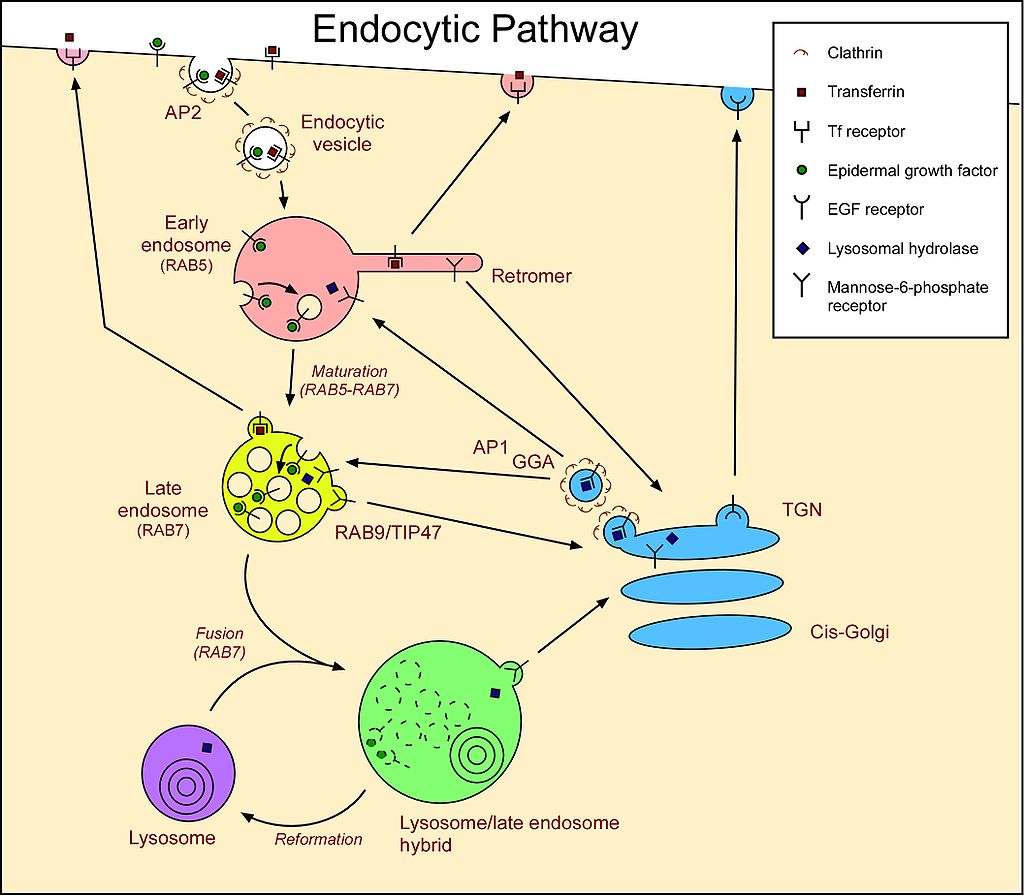
Los materiales dentro de la célula programada para la digestión se depositan primero dentro de los lisosomas. Estos pueden ser:
- otros orgánulos, como las mitocondrias, que han dejado de funcionar correctamente y han sido engullidos en autofagosomas
- moléculas de alimentos o, en algunos casos, partículas de alimentos tomadas en la célula por endocitosis
- partículas extrañas como bacterias que son engulladas por neutrófilos
- antígenos que son absorbidos por
- células presentadoras de antígeno “profesionales” como las células dendríticas (por fagocitosis) y
- Células B (por unión a sus receptores de antígeno (BCR) seguido de endocitosis mediada por receptores.
En un momento, se pensó que los lisosomas eran los encargados de matar células programadas para ser retiradas de un tejido; por ejemplo, la reabsorción de su cola a medida que el renacuajo se metamorfosis en una rana. Esto es incorrecto. Estos ejemplos de muerte celular programada (PCD) o apoptosis tienen lugar por un mecanismo completamente diferente.
En algunas células, los lisosomas tienen una función secretora —liberando su contenido por exocitosis.
- Las células T citotóxicas (CTL) secretan perforina de los lisosomas.
- Los mastocitos secretan algunos de sus muchos mediadores de inflamación a partir de lisosomas modificados.
- Los melanocitos secretan melanina a partir de lisosomas modificados.
- La exocitosis de los lisosomas proporciona la membrana adicional necesaria para sellar rápidamente las heridas en la membrana plasmática.
Las enfermedades de almacenamiento lisosómico son causadas por la acumulación de macromoléculas (proteínas, polisacáridos, lípidos) en los lisosomas debido a una falla genética en la fabricación de una enzima necesaria para su descomposición. Las neuronas del sistema nervioso central son particularmente susceptibles al daño. La mayoría de estas enfermedades son causadas por la herencia de dos alelos defectuosos del gen que codifica una de las enzimas hidrolíticas. Los ejemplos incluyen:
- La enfermedad de Tay-Sachs y la enfermedad de Gaucher, ambas causadas por la falta de producción de una enzima necesaria para descomponer los esfingolípidos (derivados de ácidos grasos que se encuentran en todas las membranas celulares).
- Mucopolisacaridosis I (MPS-I). Causada por la falta de síntesis de una enzima (α- L-iduronidasa) necesaria para descomponer los proteoglicanos como el sulfato de heparano. En abril de 2003, la Administración de Alimentos y Medicamentos de Estados Unidos aprobó una versión sintética de la enzima, laronidasa (Aldurazyme®), como posible tratamiento. Esta enzima (que contiene 628 aminoácidos) es fabricada por tecnología de ADN recombinante.
Sin embargo, una enfermedad de almacenamiento lisosómico, la enfermedad de células I (“enfermedad de células de inclusión”), es causada por la falta de “marcar” (por fosforilación) todas las enzimas hidrolíticas que se supone que deben ser transportadas desde el aparato de Golgi a los lisosomas. Al carecer de la etiqueta manosa 6-fosfato (M6P), se secretan de la célula en su lugar. El resultado: todas las macromoléculas incorporadas en los lisosomas permanecen sin degradar formando “cuerpos de inclusión” en la célula.
Peroxisomas
Los peroxisomas, también llamados microcuerpos, tienen aproximadamente el tamaño de los lisosomas (0.5—1.5 µm) y al igual que ellos están encerrados por una sola membrana. También se asemejan a los lisosomas al estar llenos de enzimas. Sin embargo, los peroxisomas brotan del retículo endoplásmico, no del aparato de Golgi (fuente de los lisosomas) y las enzimas y otras proteínas destinadas a los peroxisomas se sintetizan en el citosol. Cada uno contiene un p eroxisomal t argeting s ignal (PTS) que se une a una molécula receptora que lleva la proteína al peroxisoma y luego regresa para otra carga. Se han identificado dos señales de diana peroxisomales: una secuencia de 9 aminoácidos en el extremo N-terminal de la proteína y un tripéptido en el C-terminal. Cada uno tiene su propio receptor para llevarlo al peroxisoma.
Funciones de los peroxisomas en el hígado humano incluyen:
- Desglose (por oxidación) del exceso de ácidos grasos.
- Desglose del peróxido de hidrógeno (H 2 O 2), un producto potencialmente peligroso de la oxidación de ácidos grasos. Es catalizado por la enzima catalasa.
- Participa en la síntesis del colesterol. Una de las enzimas involucradas, la HMG-CoA reductasa, es el objetivo de las populares “estatinas” reductoras del colesterol.
- Participa en la síntesis de ácidos biliares.
- Participa en la síntesis de los lípidos utilizados para elaborar mielina.
- Desglose del exceso de purinas (AMP, GMP) a ácido úrico.
Los peroxisomas también están presentes en las células vegetales donde participan es funciones tales como la fijación simbiótica de nitrógeno y la fotorespiración.
Una variedad de trastornos hereditarios raros de la función del peroxisoma ocurren en humanos. La mayoría involucra versiones mutantes de una u otra de las enzimas que se encuentran dentro de los peroxisomas. Por ejemplo: la adrenoleucodistrofia ligada al X (X-ALD) resulta de una falla en la metabolización adecuada de los ácidos grasos. Un resultado es el deterioro de las vainas de mielina de las neuronas. El trastorno ocurre en niños pequeños debido a que el gen está ligado al cromosoma X. Un intento de encontrar un tratamiento efectivo fue el tema de la película de 1992 Lorenzo's Oil. Algunas enfermedades son el resultado de no producir peroxisomas funcionales. Por ejemplo: El síndrome de Zellweger resulta de la herencia de dos genes mutantes para uno de los receptores (PXR1) necesarios para importar proteínas al peroxisoma.