
4.1: Purificación de proteínas
- Page ID
- 57318

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Ensayos, Actividad Específica, Fraccionamiento Inicial
Un procedimiento exitoso de purificación de proteínas puede ser nada menos que increíble. Ya sea que esté comenzando con una proteína recombinante que se produce en E. coli, o tratando de aislar una proteína de algún tejido de mamífero, normalmente está comenzando con cantidades en gramos de una mezcla compleja de proteínas, ácidos nucleicos, polisacáridos, etc. de la que puede tener que extraer miligramo (¡o microgramo!) cantidades de proteína deseada a alta pureza, y ojalá con alto rendimiento.
El primer paso en cualquier purificación es el desarrollo de un ensayo específico para la proteína de interés. El ensayo específico puede basarse en alguna característica única de la proteína de interés
- Actividad enzimática
- Actividad inmunológica
- Características físicas (por ejemplo, masa molecular, propiedades espectroscópicas, etc.)
- Actividad biológica
- Idealmente, un ensayo debería ser
- Específico (no quieres un falso positivo)
- rápido (no quieres esperar una semana por los resultados)
- sensible (no quieres consumir toda tu muestra para analizarla)
- cuantitativo (necesita una manera precisa de medir la cantidad de su proteína en cada paso de la purificación)
Western Blotting
Los anticuerpos pueden ser utilizados en un método llamado Western blotting, que es útil para determinar los niveles de expresión de proteínas y para ensayar proteínas durante la purificación. Este método generalmente implica los siguientes pasos:
- Una muestra de proteína se somete a electroforesis en gel de poliacrilamida.
- Después de esto, el gel se coloca sobre una lámina de nitrocelulosa y la proteína en el gel se transfiere electroforéticamente a la nitrocelulosa.
- Luego, la nitrocelulosa se remoja en gelatina para “bloquear” su capacidad de unirse de manera no específica a las proteínas.
- Luego se incuba la nitrocelulosa con el anticuerpo específico para la proteína de interés.
- La nitrocelulosa se incuba luego con un segundo anticuerpo que es específico para el primer anticuerpo. Por ejemplo, si el primer anticuerpo se crió en conejos, el segundo anticuerpo podría denominarse “inmunoglobulina anti-conejo de cabra”. Lo que esto significa es que se utilizaron inmunoglobulinas de conejo para provocar una respuesta de anticuerpos en cabras. Los anticuerpos de cabra (policlonales) incluirán aquellos que reconocen la región conservada en los anticuerpos de conejo. Dado que la región Fc está conservada, se unirá a todos y cada uno de los anticuerpos de conejo, incluidos los que están en el papel de nitrocelulosa.
- El segundo anticuerpo típicamente tendrá una enzima unida covalentemente que, cuando se proporciona con un sustrato cromogénico, provocará una reacción de color.
- Así, el peso molecular y la cantidad de la proteína deseada se pueden caracterizar a partir de una mezcla compleja (por ejemplo, extracto celular crudo) de otras proteínas.
En una variación de lo anterior, la muestra de proteína puede ser transferida directamente sobre un papel de nitrocelulosa (llamado dot blot) sin primero correr un gel. Esto puede ser deseable si, por ejemplo, el anticuerpo es monoclonal y reconoce un epítopo que depende de la estructura nativa (que se destruiría al ejecutar una SDS PAGE).
Además de sus variados usos, los anticuerpos también pueden ser utilizados para purificar proteínas.
- Si se pueden obtener cantidades relativamente grandes de un anticuerpo, se pueden unir covalentemente a una resina de cromatografía (por ejemplo, perlas de Sephadex).
- Si un extracto celular crudo se pasa sobre una columna de este tipo, solo la proteína de interés debe unirse, y todo lo demás fluirá a través de ella.
- La proteína unida puede entonces eluirse. Esto se logra típicamente mediante condiciones de pH moderadamente bajas (usando ácido acético). Mientras la proteína de interés no se desnaturalice irreversiblemente por tales condiciones, el método funcionará bastante bien.
- Un problema potencial implica el de los anticuerpos monoclonales que se utilizan para purificar proteínas mutantes. Las regiones de la proteína que comprende el epítopo no pueden modificarse sin destruir la capacidad del anticuerpo para unirse. Así, el uso de anticuerpos monoclonales en un esquema de purificación puede impedir su uso en la purificación de ciertos mutantes.
La purificación de proteínas puede considerarse como una serie de etapas de fraccionamiento diseñadas de manera que:
- La proteína de interés se encuentra casi exclusivamente en una fracción (y con buen rendimiento)
- Una cantidad significativa de los contaminantes se puede encontrar en una fracción diferente
Durante la purificación, deberá monitorear varios parámetros, entre ellos:
- Volumen total de la muestra
- Proteína de muestra total (puede estimarse por A 280; 1.4 ~ 1.0 mg/ml)
- Unidades de actividad de la proteína deseada (basado en ensayo específico)
Esta información básica le permitirá realizar un seguimiento de la siguiente información durante cada paso de purificación:
- % de rendimiento para cada etapa de purificación
- Actividad específica de la proteína deseada (unidades/mg de proteína total)
- Mejora de la purificación de cada etapa (por ejemplo, purificación 3.5x)
En el diseño de un esquema de purificación normalmente hay que equilibrar la purificación con el rendimiento.
- Por ejemplo, puede ser relativamente sencillo obtener 90% de material puro con buen rendimiento.
- Sin embargo, puede ser difícil mejorar esa pureza un percentil adicional con buen rendimiento.
- La aplicación planificada de la proteína purificada determina la pureza objetivo.
- Si la proteína va a ser utilizada para determinar la información de la secuencia de aminoácidos, tal vez 90% es aceptable. Sin embargo, si el material va a ser utilizado en ensayos clínicos, 99.99+% puede ser la pureza objetivo.
Pasos iniciales en la purificación
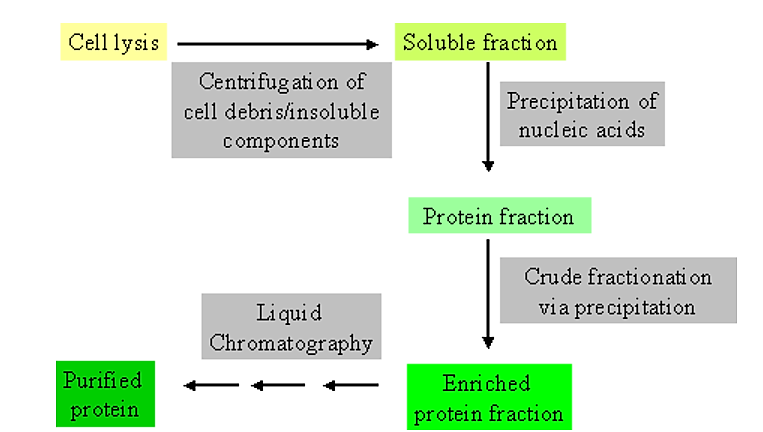.png)
Figura 4.1.1: Etapas de purificación
- Es sumamente útil tener alguna información no sólo sobre las características físicas y químicas generales de la proteína que intentas purificar, sino también sobre los componentes contaminantes.
- Por ejemplo, muchas proteínas de E. coli son generalmente de bajo peso molecular (<50,000 Da) y algo ácidas en el punto isoeléctrico
Por lo general, los pasos iniciales en la purificación hacen uso de diferencias físicas y/o químicas generales entre las proteínas solubles y otros componentes celulares.
- Por ejemplo, las proteínas solubles pueden separarse de los desechos celulares generales, y las células intactas, por centrifugación.
- Así, las células se alteran físicamente (a través de homogeneización o una prensa celular) para permitir la liberación del contenido celular. A esto le sigue la centrifugación para separar los componentes generalmente solubles de aquellos que son insolubles.
- Es en este punto que se inicia la recolección de datos con el fin de monitorear la purificación.
Los ácidos nucleicos a veces se pueden eliminar fácilmente de la muestra mediante la adición de compuestos catiónicos grandes tales como polietilenimina o sulfato de estreptomicina.
- Los ácidos nucleicos se unen a estos compuestos a través de interacciones electrostáticas y el complejo precipita y se puede eliminar mediante centrifugación.
- El mismo resultado general se puede obtener mezclando en resinas de intercambio iónico que son intercambiadores de aniones (es decir, las resinas contienen grupos catiónicos) y luego filtrando o centrifugando para eliminar. Al igual que con cualquiera de los dos métodos, se debe confirmar que la proteína deseada no está unida también.
Los fraccionamientos crudos de proteínas se pueden lograr mediante la adición de diversas cantidades de precipitantes como sulfato de amonio, o polietilenglicol (PEG).
- Para este tipo de etapa de purificación se realiza un experimento inicial para monitorear la fracción de proteína global, así como la proteína deseada, permaneciendo en solución (y pellet) en función de la concentración de precipitante.
|
Sulfato de Amonio (% saturado) |
0 |
10 |
20 |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
|
Muestra A 280 |
1000 |
900 |
600 |
200 |
100 |
75 |
50 |
40 |
25 |
20 |
|
Ensayo de actividad (unidades) |
200 |
200 |
200 |
190 |
170 |
100 |
30 |
5 |
0 |
0 |
.png)
Figura 4.1.2: Actividad proteica en función de la concentración de precipitante
- En este ejemplo en particular estamos de suerte: en alrededor del 30% de sulfato de amonio podemos precipitar alrededor del 80% de la concentración total de proteína en nuestra muestra, sin embargo, nuestro ensayo de actividad para nuestra proteína deseada indica que alrededor del 95% de nuestra proteína deseada sigue siendo soluble.
- Al 80% de sulfato amónico toda nuestra proteína deseada ha precipitado. Así, a partir de estos resultados haríamos lo siguiente:
- Agregar sulfato de amonio a nuestra muestra a una concentración de 30% de saturación
- Centrifugar y desechar el pellet
- Añadir sulfato de amonio al 80% de saturación
- Centrifugar y mantener el pellet. Resuspender el sedimento en tampón para solubilizar la proteína.
- Se esperaría una purificación de aproximadamente 5 veces con aproximadamente 95% de rendimiento.
Cromatografía en Columna - Intercambio iónico; Diálisis y concentración
Cromatografía en columna
Después de las etapas iniciales de fraccionamiento el procedimiento típico es pasar a cromatografía en columna.
- En la cromatografía en columna tenemos un tubo de vidrio (columna) que se llena con un material (“resina”) que tiene ciertas características físicas/químicas.
- Estas características le permiten interactuar de diversas maneras con diferentes proteínas.
- Algunos tipos comunes de resinas cromatográficas incluyen:
- Intercambio iónico
- Hidrofóbico
- Filtración en gel
- Afinidad
Intercambio iónico
Las resinas de intercambio iónico contienen grupos cargados.
- Estos pueden ser de naturaleza ácida (en cuyo caso la resina es un intercambiador catiónico)
- o básico (en cuyo caso es un intercambiador aniónico).
- Los intercambiadores catiónicos y aniónicos se pueden descomponer aún más en intercambiadores débiles y fuertes (reflejando la afinidad de unión).
|
Tipo de intercambiador |
Grupo funcional |
Nombre común |
|---|---|---|
|
Intercambiador de cationes débiles |
carboximetilo |
CM celulosa/sephadex |
|
Intercambiador de cationes fuerte |
sulfopropilo |
Sephadex SP |
|
Intercambiador aniónico débil |
dietiaminoetilo |
DE celulosa/sephadex |
|
Intercambiador de aniones fuerte |
amina cuaternaria |
Sephadex QAE |
- Por lo general, las muestras se cargan en condiciones de baja fuerza iónica y el material unido se eluye usando una etapa o gradiente de elución de tampón con mayor fuerza iónica.
- En términos generales, una proteína se unirá a una resina de intercambio catiónico si el pH del tampón es menor que el punto isoeléctrico (pI) de la proteína, y se unirá a una resina de intercambio aniónico si el pH es mayor que el pI.
.png)
Figura 4.1.3: Unión de proteínas a resinas
- Por lo tanto, el conocimiento del pI de la proteína es útil para diseñar un protocolo de purificación utilizando resinas de intercambio iónico (sin embargo, siempre puedes probar diferentes resinas para ver cuál funciona mejor).
En términos generales, las columnas de intercambio iónico son cortas y gordas en dimensiones.
Elución de proteínas de resinas de intercambio iónico
- Las proteínas unidas a resinas de intercambio iónico se unen a través de interacciones iónicas no covalentes (puente salino). Podemos competir por estos sitios de unión iónica en la resina con otros grupos iónicos, a saber, sales
- Existen dos tipos generales de métodos al eluir con una solución salina: 1. Gradiente de elución y 2. Elución por etapas
- Una elución en gradiente se refiere a una transición suave de la concentración de sal (de baja a alta) en el tampón de elución. Las proteínas de unión débil eluyen primero, y las proteínas de unión más fuertes eluyen en último lugar (es decir, requieren mayores concentraciones de sal en el tampón para competir con ellas fuera de la columna)
- Se puede hacer una concentración de sal en gradiente usando un fabricante de gradientes. En su forma más simple, esta consiste en dos contenedores (deben ser de la misma forma) conectados por un sifón (o tubo en la parte inferior). Un recipiente contiene el tampón bajo en sal y el otro contiene tampón alto en sal. El tampón se retira del recipiente bajo en sal:
.png)
Figura 4.1.4: Creador de gradientes
- Esto producirá un gradiente lineal de concentraciones bajas a altas de sal sobre el volumen total del gradiente
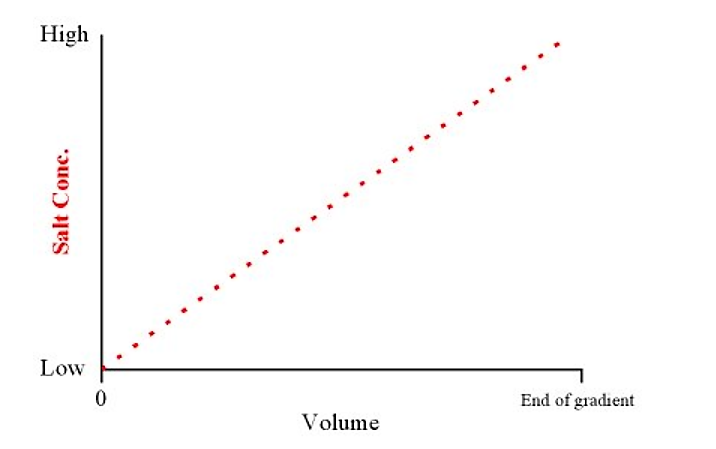.png)
Figura 4.1.5: Concentración y volumen de sal
- Si conocemos el rango de concentración de sal sobre el cual eluirá una proteína de interés, simplemente podemos eluir con un tampón que contenga esa concentración de sal. Esto se conoce como elución escalonada.
- Las eluciones escalonadas son generalmente más rápidas de ejecutar y eluyen la proteína en un volumen global más pequeño que con las eluciones en gradiente. Por lo general, funcionan mejor cuando los contaminantes eluyen a una concentración de sal significativamente diferente a la proteína de interés
.png)
Figura 4.1.6: Elución escalonada
Tenga en cuenta que después de la cromatografía de intercambio iónico la proteína de interés estará en un tampón con una concentración potencialmente alta de sal. Esto debe tenerse en cuenta antes de continuar con el siguiente paso en el esquema de purificación
Diálisis
- Después de una etapa de precipitación con sulfato de amonio, o una etapa de cromatografía de intercambio iónico, la proteína de interés puede estar en un tampón alto en sal. Esto puede ser indeseable por varias razones. ¿Cómo nos deshacemos de la sal en nuestra muestra?
- Uno de los métodos más comunes es el de la diálisis
- El método de diálisis hace uso de membranas semipermeables. En el ejemplo más simple, esta membrana se fabrica en forma de tubo (que se parece mucho a una envoltura de salchicha)
- La característica principal de esta membrana es que es porosa. Sin embargo, el tamaño de poro es tal que aunque los iones de sal pequeños pueden pasar libremente a través de la membrana, las moléculas de proteína más grandes no pueden (es decir, se retienen) Así, las membranas de diálisis se caracterizan por la masa molecular de la proteína globular típica más pequeña que retendrá.
- Esto se conoce comúnmente como el corte del tubo (por ejemplo, el tubo de diálisis Spectrapore #6 tiene un corte de 1,000 Daltons, lo que significa que una proteína de 1,000 Dalton será retenida por el tubo pero que los solutos de masa molecular más pequeños pasarán a través del tubo)
- La diálisis procede colocando una muestra con alto contenido de sal en tubos de diálisis (es decir, la “bolsa” de diálisis) y colocándola en el tampón bajo en sal deseado:
.png)
Figura 4.1.7: Diálisis
- Con el tiempo la concentración de solutos de baja masa molecular dentro de la bolsa, y en el tampón bajo en sal, llegará a alcanzar el equilibrio. En términos prácticos (para el caso anterior) las moléculas de sal se difundirán fuera de la bolsa hacia el tampón bajo en sal:
.png)
Figura 4.1.8: Difusión de sal
- En equilibrio, la concentración de sal de la muestra se puede calcular de la siguiente manera:
$$\ frac {(muestra\: volumen)\ veces (muestra\: sal\: concentración) + (buffer\: volumen)\ veces (buffer\: sal\: concentración)} {total\ :volumen} = final\ :sal\: concentración$$
Nota
A menudo, la concentración de sal tampón es de 0 M
- El volumen de tampón para la diálisis es una función de la concentración final requerida de sal en la muestra
Ejemplo 4.1.1:
Ejemplo de diálisis
Tenemos una muestra de proteína de 10 ml de un conjunto de elución de columna de intercambio iónico que contiene NaCl 1.0M. Para nuestro siguiente paso en la purificación no podemos tener más de 1 mM de NaCl en la muestra.
.png)
Por lo tanto, el volumen de tampón requerido sería (vol total - volumen de muestra) = 9.990 L (o ~ 10 L)
- Así, si dializamos 10mls de muestra (con 1.0M NaCl conc) en 10 L de agua después del equilibrio la concentración de NaCl en la muestra sería 1.0 mM.
- Tenga en cuenta que en el ejemplo anterior esto comúnmente se denominaría diálisis "1:1 ,000".
- ¿Supongamos que no queremos componer 10 L de buffer? De hecho, podemos lograr los mismos resultados con dos dializaciones secuenciales "1:32" (es decir, la raíz cuadrada de la diálisis 1:1 ,000, es decir, dos dializaciones secuenciales 1:32 equivalen a una única diálisis 1:1 ,000):
Primera diálisis versus 310 ml de tampón: la concentración de NaCl de la muestra será (10*1.0)/(320) = 31 mM
Segunda diálisis versus 310 ml de tampón: la concentración de NaCl de la muestra será (10*0.031)/(320) = 0.97 mM
Por lo tanto, en lugar de hacer 10 L de tampón, podríamos hacer solo 620 ml y lograr los mismos resultados con dos etapas de diálisis
- En este caso, retirar la sal tomaría el doble de tiempo, es decir, necesitamos realizar dos etapas de diálisis. ¿Cuánto dura la diálisis?
Una regla general útil es que para la mayoría de los tipos de tubos de diálisis la diálisis es 80% compite después de cuatro horas
- Una consecuencia de la diálisis a tener en cuenta es que mientras los iones de sal se mueven fuera de la bolsa, las moléculas de agua se están moviendo dentro de la bolsa. Así, el volumen de muestra puede aumentar realmente (la bolsa se hinchará) y, por lo tanto, la concentración de proteína disminuirá
- En el caso extremo, la bolsa en realidad puede hincharse hasta el punto de ruptura. Por lo tanto, es una buena idea no llenar la bolsa por completo, sino dejar un vacío para permitir una posible hinchazón.
Concentración
- ¿Y si nuestra muestra de proteína está demasiado diluida para nuestras necesidades? ¿Cómo podemos concentrar nuestras muestras?
- Un método común es, nuevamente, utilizar una membrana semipermeable para este propósito.
- Un método muy sencillo es colocar nuestra muestra en una bolsa de diálisis y cubrirla con un soluto de alto peso molecular que puede ser fácilmente disuelto por el tampón.
- Por ejemplo, los polietilen glicoles y las polivinil pirrolidonas pueden tener masas moleculares muy grandes (es decir, 20,000 Da). Estos compuestos también se disuelven fácilmente en agua. Si nuestra muestra en una bolsa de diálisis está recubierta con formas secas de los polímeros anteriores, el agua saldrá de la bolsa de diálisis (puede pasar por los poros) e hidratar los polímeros. El resultado es una disminución en el volumen de tampón en la bolsa de diálisis (la proteína se concentrará).
- En otra variación, la membrana semipermeable se fabrica en un disco plano y se coloca en el fondo de un recipiente que contiene nuestra muestra. En un método el recipiente se presuriza y fuerza el tampón fuera del recipiente (la proteína se retiene y se concentra). En otro método, el vaso se centrifuga y la fuerza centrípeta logra el mismo objetivo que la presión en el ejemplo anterior.
Tanto para diálisis como para concentración, es esencial que la membrana no interactúe con la proteína (es decir, no tenga afinidad por, y no se unirá a, la proteína)
- Con los concentradores de tipo presión, se puede lograr diálisis y concentración en tándem. Por ejemplo, la muestra puede concentrarse y luego agregarse tampón a la muestra. A continuación, la muestra se concentra de nuevo. Cada vez que se agrega tampón se reduce la concentración de sal. Después de ciclos repetidos de esto, la concentración de sal se encuentra en el nivel deseado y la muestra se concentra hasta el volumen final deseado.
Filtración en Gel, Afinidad y Resinas Hidrofóbicas; Preparación de Resina,
Filtración en gel
La filtración en gel no se basa en ninguna interacción química con la proteína, más bien se basa en una propiedad física de la proteína, que es el radio molecular efectivo (que se relaciona con la masa para la mayoría de las proteínas globulares típicas).
- La resina de filtración en gel se puede considerar como perlas que contienen poros de un rango de tamaño definido.
- Las proteínas grandes que no pueden entrar en estos poros pasan alrededor del exterior de las perlas.
- Las proteínas más pequeñas que pueden entrar en los poros de las perlas tienen un camino más largo y tortuoso antes de que salgan de la perla.
- Así, una muestra de proteínas que pasan por una columna de filtración en gel se separará en función del tamaño molecular: las grandes eluirán primero y las más pequeñas eluirán las últimas (y las proteínas de tamaño “medio” eluirán en el medio).
.png)
Figura 4.1.9: Filtración en gel
- Si su proteína es inusualmente “pequeña” o “grande” en comparación con las proteínas contaminantes, entonces la filtración en gel puede funcionar bastante bien.
¿Dónde se eluirá una proteína en un experimento de filtración en gel?
- Hay dos extremos en el perfil de separación de una columna de filtración en gel.
- Existe una masa molecular crítica (masa grande) que quedará completamente excluida de las perlas de filtración en gel. Todos los solutos de la muestra que sean iguales o mayores que este tamaño crítico se comportarán de manera idéntica: todos se eluirán en el volumen excluido de la columna
- Existe una masa molecular crítica (masa pequeña) que se incluirá completamente dentro de los poros de las perlas de filtración en gel. Todos los solutos de la muestra que sean iguales o menores que este tamaño crítico se comportarán de manera idéntica: todos se eluirán en el volumen incluido de la columna
- Los solutos entre estos dos rangos de masa molecular eluirán entre los volúmenes excluidos e incluidos
.png)
Figura 4.1.10: Volumen excluido vs. incluido
Como regla general, el volumen excluido (Vo) es aproximadamente igual a un tercio del volumen de columna, el volumen incluido es aproximadamente igual a dos tercios del volumen de columna
- En la filtración en gel la resolución es una función de la longitud de la columna (cuanto más larga, mejor)
- Sin embargo, un inconveniente está relacionado con el volumen máximo de muestra que se puede cargar. Cuanto mayor sea el volumen de muestra cargado, mayor será el solapamiento entre los picos separados. En términos generales, el tamaño de la muestra que se puede cargar se limita a aproximadamente 3-5% del volumen total de la columna.
- Por lo tanto, la filtración en gel se guarda mejor para las etapas finales de una purificación, cuando la muestra se puede concentrar fácilmente a un pequeño volumen.
- La filtración en gel también se puede utilizar para eliminar sales de la muestra, debido a su capacidad para separar componentes “pequeños” de “grandes”.
- Finalmente, la filtración en gel puede estar entre los métodos de purificación más “suaves” debido a la falta de interacción química con la resina.
Cromatografía de afinidad
La cromatografía de afinidad es un término general que se aplica a una amplia gama de medios cromatográficos. Básicamente puede pensarse como alguna resina inerte a la que se le ha unido algún compuesto que tenga una afinidad específica por su proteína de interés.
- Así, un anticuerpo específico unido a una resina inerte sería un tipo de cromatografía de afinidad.
- Otros ejemplos podrían incluir: un inhibidor de proteasa unido a alguna matriz, diseñado para unirse a una proteasa específica
- un cofactor unido a alguna matriz, diseñado para unirse a una enzima particular
- un ion metálico unido a una matriz, diseñado para quelar una proteína con un sitio de unión a metal, y así sucesivamente.
En cada caso, el tipo de resinas utilizadas y el método de unión pueden variar, al igual que el método de elución. Una generalización con respecto al método de elución es que el ligando unido puede competir fuera del grupo funcional de la columna incluyendo en el tampón de elución una alta concentración del grupo funcional libre. Por ejemplo, si el grupo funcional de la columna es un cofactor, entonces la proteína unida puede competir fuera de la columna pasando un tampón que contiene una alta concentración de cofactor (o análogo de cofactor) a través de la columna.
Otros métodos de elución incluyen cambiar las condiciones del tampón de manera que la proteína ya no esté en el estado nativo (ya que es el estado nativo el que confiere la estructura requerida para la interacción de unión específica). Esto se puede lograr cambiando el pH o añadiendo agentes desnaturalizantes como urea o guanidina.
Con la cromatografía de afinidad, típicamente la purificación lograda en una sola etapa puede ser dramática, del orden de varios miles de veces. Las purificaciones de un solo paso con columnas de afinidad específicas no son inauditas, de hecho es un objetivo ideal de purificación, una matriz que reconoce solo la proteína de interés y ninguna otra.
Resinas hidrófobos
Las resinas hidrofóbicas contienen un grupo funcional no polar, tal como un alcano o un grupo aromático.
- “Muchas proteínas son capaces de secuestrar dichos grupos en su superficie y esta exclusión del disolvente proporciona la base de la energía de unión (es decir, el “" efecto hidrófobo "”).”
- Esta interacción se potencia al aumentar la fuerza iónica, de tal manera que las proteínas pueden unirse en condiciones de alto contenido de sal y eluir en condiciones bajas en sal.
- Como tales, estas columnas pueden usarse no solo para proporcionar purificación, sino para desalar muestras (por ejemplo, después de una precipitación inicial con sulfato de amonio).
- Por lo general, no es posible predecir de antemano qué resina particular se unirá a una proteína dada, esto generalmente se determina empíricamente. Sin embargo, cuanto más largo sea el alcano, o cuanto mayor sea el compuesto aromático, más fuerte será la unión típicamente.
Debido a la naturaleza de las interacciones hidrófobas y la fuerza iónica, la cromatografía hidrofóbica y la cromatografía de intercambio iónico pueden usarse convenientemente secuencialmente. Por ejemplo, después del intercambio iónico la proteína se encuentra en condiciones de alto contenido de sal, por lo que se puede cargar directamente en una columna hidrofóbica. Por el contrario, una columna hidrofóbica se eluye en baja sal, lo que es un requisito para unirse a una resina de intercambio iónico.
Se debe señalar una distinción entre la cromatorgrafía de interacción hidrófoba y la cromatografía de fase inversa
- La cromatografía de interacción hidrofóbica se realiza en condiciones de disolvente acuoso y se utilizan cambios en la fuerza iónica para eluir la columna. La proteína típicamente se une en estado nativo a través de grupos hidrófobos ubicados en la superficie de la proteína. El estado nativo se conserva durante las condiciones de elución
- La cromatografía de fase inversa utiliza un disolvente hidrófobo (típicamente acetonitrilo) y la unión de un ligando es una función de la división de fases entre la naturaleza hidrófoba del disolvente y el grupo funcional de la columna. Las proteínas se desnaturalizan típicamente en tales disolventes y se unen debido a la naturaleza hidrófoba de la secuencia polipeptídica completa. Dado que la mayoría de los grupos hidrófobos están localizados en el núcleo de las proteínas globulares, la unión está relacionada con la desnaturalización de la proteína y la accesibilidad de estos grupos a los grupos funcionales de la columna. Las proteínas pueden purificarse usando cromatografía de fase inversa, pero generalmente deben replegarse de alguna manera para recuperar la funcionalidad (es decir, el estado nativo)
Preparación de resinas
Las etapas en la preparación de una resina cromatográfica implican típicamente:
- Hidratación de resina
- Multas de decantación
- Equilibrar la resina y preparar una suspensión
- Desgasificación de la lechada
- Las resinas vienen secas o prewollen. Si están secos necesitan estar hidratados. Esto generalmente se logra mezclando la resina seca con tampón y dejándola hidratar lentamente durante la noche (o más rápido a temperaturas más altas
- Después de que la resina se haya hidratado y asentado, partículas muy finas se asentarán en la parte superior. Estos “finos” ralentizan el caudal de la resina empaquetada. Por lo tanto, la resina sedimentada se decanta cuidadosamente para desechar estos finos.
- A continuación, la resina se equilibre en el tampón que se utilizará para el análisis. El equilibrio generalmente implica pH de la resina, o intercambios de tampón. Nunca use una barra de agitación cuando aplique el pH de la resina (puede cizallar mecánicamente la resina y producir finos), más bien revuelva la suspensión de resina con una varilla de agitación.
- Después de que la resina equilibrada se haya asentado, se agrega un volumen igual de tampón para producir una suspensión de resina al 50%. Esto suele ser lo suficientemente “delgado” como para permitir que las burbujas de aire escapen al empaquetar la columna.
- Finalmente, la suspensión se desgasifica antes de empaquetar la columna. Esto ayudará a minimizar la formación de burbujas de aire.
Empaque de la columna
Las columnas de baja presión se empaquetan típicamente usando gravedad.
- Agrega una pequeña cantidad de búfer al fondo de la columna.
- Coloque un depósito de empaque en la parte superior de la columna. Ya que estaremos usando una suspensión al 50% tendremos un volumen que es 2x el volumen de la columna y lo mejor es verter la resina en todo a la vez. Por lo tanto, el depósito de empaque debe tener un volumen igual o mayor que el volumen de la columna.
- Vierta cuidadosamente la suspensión de resina en el depósito/columna de empaque, evitando la introducción de burbujas de aire tanto como sea posible
- Deje que la columna repose durante aproximadamente 5 minutos para permitir que escapen grandes burbujas de aire
- Abra la válvula de la columna en la parte inferior y permita que la columna se empaque bajo gravedad
- Tenga en cuenta la parte superior del lecho de resina. Se moverá hacia abajo a medida que la columna se empaca. Cuando la columna está empaquetada, la parte superior del lecho de resina ya no se moverá hacia abajo.
Plomería
Los sistemas de cromatografía se pueden ejecutar usando solo gravedad y un vaso de precipitados para recoger la fracción apropiada. Sin embargo, los sistemas más comunes incluirán los siguientes:
- Una bomba. Por lo general, una bomba peristáltica con caudal variable y un puerto de comunicaciones para un controlador. La bomba generalmente se configura para empujar el tampón a través de la columna, en lugar de succionar el tampón de la columna (lo que puede causar una condición de baja presión con la producción de burbujas de aire)
- Un detector. Este es típicamente un detector UV (A 280). La mayoría de los detectores son del tipo de dos celdas, lo que significa que puede tener un búfer simple como blanco en el detector mientras analiza sus fracciones de columna. El detector envía la información de absorbancia a un registrador gráfico para ser visualizado (impreso)
- Un colector de fracciones. Esto permite recolectar fracciones ya sea por número de gotas (~30 por ml) o por tiempo. En conjunto con una bomba controlable, la recolección de tiempo se traduce en volumen. El colector de fracciones normalmente tendrá un puerto de comunicaciones para emitir una señal cuando cambia de fracción y para recibir comandos del detector/controlador en algunos sistemas sofisticados.
- Un registrador de gráficos. Esto imprimirá una traza continua de la salida del detector y el marcador de eventos del colector de fracciones (señalización cuando una fracción cambia). Las fracciones también se pueden leer individualmente en un espectrofotómetro UV si un registrador gráfico no está disponible.
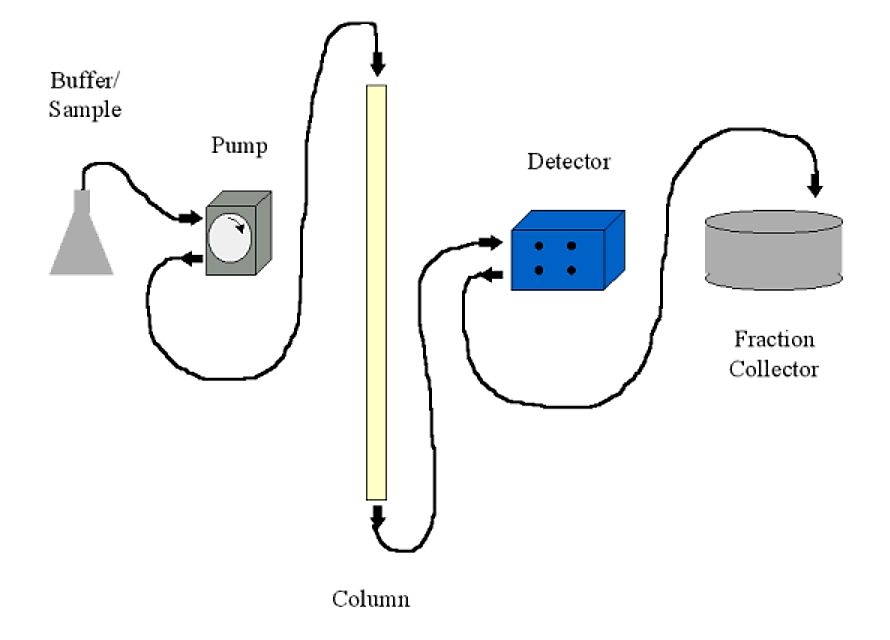.png)
Figura 4.1.11: Configuración de cromatografía
- Si se utiliza un sistema de gravedad, se debe instalar un bucle de seguridad para evitar que la columna se seque si el amortiguador se agota cuando la columna está desatendida
.png)
Figura 4.1.12: Bucle de seguridad
Tenga en cuenta que la parte inferior del bucle de seguridad es más baja que la salida al colector de fracciones.
Ejecución del experimento, resolución de picos
A continuación se presenta un ejemplo de un experimento de cromatografía líquida de baja presión (resina de intercambio iónico).
Muestra:
- Volumen = 90 mls
- A 280 = 1.8
- Total A 280 = 162
Columna:
- DE-52 (dietilaminoetil celulosa; intercambio aniónico)
- Tamaño = 1.0 x 12.7 cm
- Volumen =
.png) = 40 mL
= 40 mL
Colector de fracciones:
- 10 mls/fracción (~300 gotas/fracción)
El cromatograma para este experimento se veía así:
.png)
Figura 4.1.13: Cromatograma
Durante esta operación de cromatografía tuvieron lugar los siguientes eventos:
1. Anote las marcas de verificación en el cromatograma.
- El marcador de “evento” del colector de fracciones notifica al registrador de gráficos cuando se produce un cambio de tubo.
- El experimento comienza con la garrapata junto al '0' en el eje x (” tick 0 “); esto indica el inicio de la fracción (tubo) número 1.
- La siguiente marca de verificación (“tick 1") indica el final de la fracción número 1, y el inicio de la fracción número 2.
- Así, las fracciones abarcan la brecha entre las marcas de verificación.
2. La carga de la muestra se inicia en la garrapata 0.
3. En algún momento durante la fracción 5 comenzamos a notar que la absorbencia del efluente de la columna aumenta
- Se han tomado aproximadamente (5 fracciones x 10mls por fracción) o 50 mls desde el inicio de la carga hasta que el detector anota alguna absorbancia.
- Esto se compara bien con el hecho de que el volumen de la columna es de aproximadamente 40 mls y hay algún volumen asociado con el tubo que entra y sale de la columna.
- Por lo tanto, este 'retraso' de la carga de la muestra a la detección de la muestra es el volumen muerto del sistema
4. Obviamente, algún material no se une a la resina durante la etapa de carga. Este es el flujo a través. ¿Es éste algún componente de la muestra que no tiene afinidad por la resina, o bien, representa que hemos superado la capacidad de la resina?
- Si hemos superado la capacidad de la resina, entonces el flujo pasante tendrá una A 280 similar a la muestra que se está cargando
- Además, antes de superar la capacidad, el flujo pasante tendrá alguna característica A 280 que luego pasará a otro A 280 (el de la muestra cargada), dando como resultado un cromatograma de doble meseta.
- En el experimento anterior las mesetas de flujo pasante alrededor de A 280 = 0.5 o aproximadamente 25% de la absorbancia de la carga. Esto parecería indicar que un componente, o componente (s), que representa una cuarta parte de nuestra muestra, no tiene afinidad por la resina en la columna
5. Alrededor de la fracción 9 comenzamos a lavar la columna
- Esto tiene sentido porque se han cargado 9 fracciones x 10mls por fracción = 90 mls y esto es equivalente a nuestro volumen de muestra original (es decir, toda la muestra se ha cargado)
- La columna se lava típicamente usando las mismas condiciones de tampón en la muestra de proteína.
6. Alrededor de la fracción 14 notamos que la A 280 comienza a disminuir
- Esto tiene sentido dado que determinamos que el volumen muerto del sistema era aproximadamente 50 mls o 5 fracciones. Así, se observa que un lavado que se inició en la fracción 9 disminuye la absorbancia alrededor de la fracción 14
- Continuamos lavando la columna hasta que la A 280 se aproxima a 0 (línea base). En otras palabras, todo el material no aglutinante en la muestra ha sido lavado
7. Después de que el A 280 vuelva a la línea base comenzamos nuestro protocolo de elución. En este experimento particular utilizaremos un gradiente lineal de concentración creciente de sal (NaCl) (en tampón de lavado) para competir con el material unido a la resina de intercambio iónico.
8. Nuestra elución ha producido dos picos: un pequeño pico centrado alrededor de la fracción 42 y un pico más grande centrado alrededor de la fracción 50
- Tendremos que ensayar cada pico (y el flujo a través) para averiguar a dónde ha ido nuestra proteína de interés
- Los dos picos de elución están bastante bien resueltos. Podríamos combinar las fracciones 40-44 y llamarlo “pico 1", y combinar las fracciones 46-55 y llamarlo “pico 2".
9. ¿Queda algún material en la columna? Las áreas integradas (es decir, sumando los A 280 de cada fracción en un charco) del flujo pasante, pico 1 y pico 2 son las siguientes
- Flujo a través: 4
- Pico 1:2
- Pico 2:10
- Esto da un área total integrada de 16. Cada fracción es de 10 mls, por lo que esto da un total A 280 = 16 x 10 = 160 que es bastante cercano al total A 280 de nuestra muestra cargada.
- En otras palabras, parece que nuestro cromatograma está contabilizando todos los componentes de nuestra muestra original.
10. Si nuestra proteína de interés era realmente el pico 1 (y si nuestro rendimiento era del 100%), entonces esta columna ha proporcionado una purificación ocho veces (2 x 10/162).
Resolviendo picos
- Los picos contaminantes no necesariamente estarán completamente separados del pico que contiene nuestra proteína de interés
- En la siguiente imagen se están resolviendo dos componentes, y están presentes en cantidades equimolares (así, la pureza inicial es del 50%). El rendimiento y la pureza se listan para la situación en la que debíamos juntar cada pico dividiendo en el punto medio entre ellos (en este ejemplo particular el rendimiento y la pureza son idénticos en cada caso)
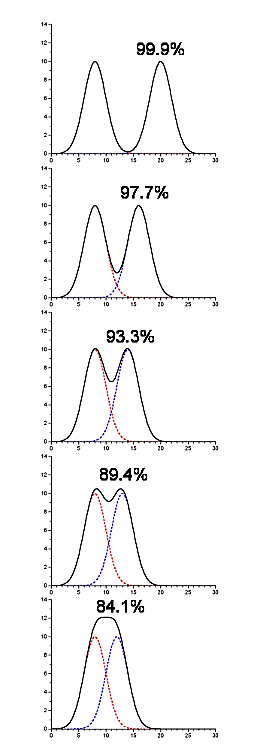.png)
Figura 4.1.14: P eaks contaminantes
- Esto le da una idea de la cantidad de contaminación cruzada en cada pico en función de su separación entre sí.
- El software para ajustar gaussianos a un cromatograma puede proporcionar este tipo de información
Reunificación para pureza versus rendimiento
Por lo general, probablemente estarás reuniendo fracciones de tal manera que se maximice la recuperación de tu proteína de interés. Sin embargo, siempre tienes la opción de agrupar para aumentar la pureza, y si tienes mucha proteína con la que trabajar esto puede permitirte lograr la pureza deseada con menos pasos. Aquí hay un ejemplo de cómo se hace:
.png)
Figura 4.1.15: Rendimiento vs. Pureza
- Todos estos son el mismo cromatograma, sin embargo, podemos agruparlos de manera diferente para obtener una mejor pureza (a expensas del rendimiento
- El pico azul es el pico de interés y no se resuelve a partir de un pico contaminante (en rojo).
- La línea vertical representa la fracción más a la izquierda que usamos para agrupar el pico (agrupamos todas las fracciones a la derecha de la línea vertical para obtener nuestra proteína de interés)
- En el último panel vemos que podemos lograr aproximadamente 98.8% de pureza si estamos dispuestos a desprendernos con la mitad de nuestra proteína!
Monitoreo de la purificación
Existen varios criterios. Uno de los criterios es que no podemos mejorar la actividad específica de nuestra muestra. Este valor se refiere a la actividad funcional de nuestra muestra en relación con la concentración total de proteína de la muestra.
- En las etapas iniciales de purificación este valor será bajo (no mucha actividad en relación con la cantidad total de proteína).
- Este valor aumentará después de cada etapa de purificación a medida que eliminemos otras proteínas de la muestra.
- En algún momento la actividad específica se estabilizará, y por definición, si es pura no podemos incrementar la actividad específica.
- Puede haber un valor publicado para la actividad específica con el que podamos comparar la nuestra.
Además, cada paso de la purificación debe ser monitoreado por electroforesis en gel.
- En las etapas iniciales de purificación probablemente veremos una variedad de bandas, de diversos pesos moleculares, en nuestro gel.
- Después de los diferentes pasos de purificación, deberíamos ver la desaparición de ciertas bandas concomitantes con la concentración creciente de una determinada banda (o bandas) que representan nuestra proteína.
- Si hemos purificado con éxito nuestra proteína (y si se trata de un solo polipéptido) debemos llegar a una actividad específica constante y una sola banda en un gel.
- Métodos analíticos como HPLC o escaneo densitómetro de un gel teñido pueden darnos una idea cuantitativa de la pureza de nuestra muestra final.
El siguiente gráfico representa los datos típicos que uno monitorizaría durante una purificación:
|
|
|
|
|
|
|
|---|---|---|---|---|---|
| Lisado celular crudo |
|
|
|
||
| 30-70% Sulfato de amonio cortado |
|
|
|
|
|
| Alberca DEAE Sephadex |
|
|
|
|
|
| Piscina CM Sephadex |
|
|
|
|
|
| Alberca de Phenyl Sepharose |
|
|
|
|
|
| Piscina de Filtración en Gel |
|
|
|
|
|
| Alberca de resina de afinidad tipo #1 |
|
|
|
|
|
| Alberca de resina de afinidad tipo #2 |
|
|
|
|
|
| Purificación total |
|
||||
| Rendimiento total (%) |
|
||||