
16: Regulación de la transcripción mediante efectos sobre las ARN polimerasas
- Page ID
- 58748

{{Template.dropdown {path:” /Genetics/ "}}}
[La Dra. Tracy Nixon hizo importantes contribuciones a este capítulo.]A. Los múltiples pasos en la iniciación y elongación por la ARN polimerasa son dianas para la regulación
La ARN polimerasa tiene que
- unirse a promotores,
- formar un complejo abierto,
- iniciar la transcripción,
- escapar del promotor,
- alargar, y
- terminar la transcripción.
Ver Figura 4.2.1.
Resumiendo mucho trabajo, sabemos que los promotores fuertes tienen alta KB, kf alta, kr baja y altas tasas de eliminación de promotores. Además, los promotores débiles tienen baja KB, baja kf, alta kr y bajas tasas de aclaramiento del promotor y los promotores moderados tienen uno o más puntos “débiles”.
Para conocer estos hechos, necesitamos datos genéticos para identificar qué macromoléculas (ADN y proteínas) interactúan en un evento de regulación específico, y para determinar qué pares de bases y residuos de aminoácidos son necesarios para ese evento de regulación. También necesitamos datos bioquímicos para describir los eventos de unión y las reacciones químicas que se ven afectadas por el evento específico de regulación. Idealmente, determinaríamos todas las constantes de velocidad directa e inversa, o constantes de equilibrio (que son una función de la relación de constantes de velocidad) si las tasas son inaccesibles. Si bien, en realidad, no podemos obtener ni tasas ni constantes de equilibrio para muchos de los escalones, algunos de los pasos son susceptibles de investigación y han demostrado ser bastante informativos sobre los mecanismos de regulación.

B. Existen métodos para medir constantes de velocidad y constantes de equilibrio, y ahora se están utilizando métodos más nuevos y más precisos
Los métodos clásicos de estudios de equilibrio y análisis de datos utilizan bajas concentraciones de enzimas y hacen suposiciones que simplifican reacciones complejas para que puedan ser tratadas por integrales definidas de ecuaciones de flujo químico. También manipulan una ecuación en una forma que se puede trazar como una función lineal, y derivan estimaciones de parámetros por valores de pendiente e intercepción
Impulsados por el éxito de la tecnología de purificación de ADN recombinante y proteínas, y por el mayor poder computacional en computadoras de escritorio, los métodos clásicos están siendo reemplazados por
- el uso de grandes cantidades de enzimas para incluirlas directamente en estudios cinéticos. En este enfoque, las enzimas se utilizan en cantidades a nivel de sustrato.
- integraciones numéricas de ecuaciones de flujo químico (Simulación Cinética)
- métodos más rigurosos basados en regresión no lineal, mínimos cuadrados (NLLS), y
- analizar datos de múltiples experimentos de diferente diseño simultáneamente (análisis global de NLLS).
Estos cambios aumentan los pasos en una reacción que puede ser examinada experimentalmente, reemplazan el conjunto limitado de mecanismos simples que pueden analizarse esencialmente por cualquier mecanismo y aumentan el conocimiento del error, permitiendo sacar conclusiones con más confianza

Recuadro 1: Las ecuaciones utilizadas en este capítulo provienen de varias fuentes diferentes que utilizan diferentes nombres para lo mismo. A continuación se enumeran algunos de estos sinónimos.
C. Enfoques experimentales para las reacciones de unión macromoleculares
Hay varios métodos disponibles para medir la cantidad de proteína que se une específicamente a una molécula de ADN. Ya los hemos encontrado como métodos para localizar sitios de unión a proteínas en el ADN, y todos son susceptibles de cuantificación. Los métodos principales incluyen la unión al filtro de nitrocelulosa, los ensayos de desplazamiento de movilidad electroforética y los ensayos de protección de DNasa.
¿Cuál Técnica Experimental es la Mejor?
- El tipo de observaciones que se pueden hacer sobre el sistema difieren para diferentes enfoques experimentales.
- Estas diferencias conducen a problemas específicos con cada técnica,
- Cada técnica depende de combinar el análisis de más de un experimento para obtener suficiente información para resolver la energía libre de unión intrínseca a partir de la energía de la cooperatividad.
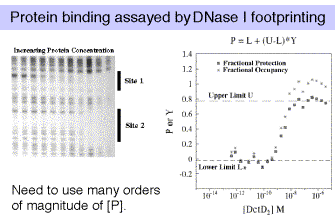
La técnica más robusta es la huella DNasa I. Si estás estudiando la unión de múltiples proteínas que interactúan, entonces es posible que estas proteínas estén mostrando cooperatividad en su unión al ADN. Al analizar dicha cooperatividad por huella de DNasa I, la resolución se limita a cooperatividades >0.5 kcal/mol, y está sujeta a algunos supuestos críticos. Los cambios en gel (también llamados ensayos de desplazamiento de movilidad electroforética, o EMSA) son útiles cuando no hay cooperatividad, o cuando la cooperatividad es grande en relación con la heterogeneidad del sitio. Los estudios de unión a filtros requieren conocimientos sobre las eficiencias de retención de filtros para los diferentes complejos de proteína-ADN, lo que solo puede determinarse empíricamente. Y siempre tenga en cuenta que las secuencias flanqueantes sí afectan las afinidades de unión, e incluso las mutaciones puntuales pueden tener efectos distantes.
En cualquiera de estos ensayos, estamos ideando un medio físico para medir una cantidad que está relacionada con la ocupación fraccionada.
D. Medición de constantes de equilibrio en reacciones de unión macromoleculares
Los métodos clásicos con su transformación lineal no son tan precisos como el análisis de regresión no lineal, mínimos cuadrados (NLLS), pero pueden servir para mostrar el enfoque general.
a.Las constantes de unión se pueden determinar valorando los sitios de unión de ADN marcados con cantidades crecientes del represor, y midiendo la cantidad de ADN unido a proteína y la cantidad de ADN libre. Las técnicas típicas son los ensayos de desplazamiento de movilidad electroforética o la unión al filtro de nitrocelulosa.
Obsérvese que para un equilibrio simple de una sola proteína que se une a un solo sitio en el ADN, la constante de equilibrio para la unión (K B) se aproxima por la inversa de la concentración de proteína a la que la concentración de ADN unido a proteína es igual a la concentración de ADN libre (Figura 4.2.3).
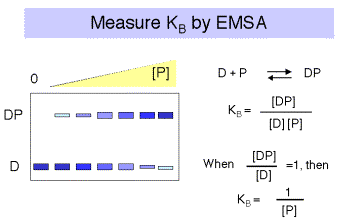
Si fuera posible determinar de manera confiable tanto la concentración de ADN unido a proteína (es decir [DP]) como la concentración de ADN libre ([D]), entonces se podría graficar la relación de ADN unido a ADN libre en cada concentración de represor. Si los resultados fueran lineales, entonces la pendiente de la línea daría la constante de unión de equilibrio, KB. Ver Figura 4.2.4.

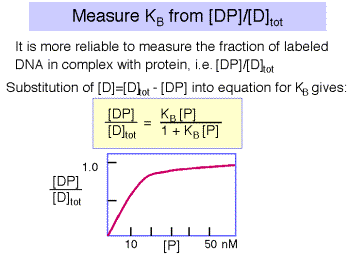
Sin embargo, el error asociado con la determinación de concentraciones muy bajas de ADN libre o unido es sustancial, y una medida más confiable es la de la relación de ADN unido a ADN total, es decir [DP]/[D] tot, como se ilustra en la Figura 4.2.5. La ecuación que describe esta curva de unión tiene una forma equivalente a la ecuación de Michelis-Menten para la cinética de enyzme en estado estacionario. Obsérvese que la concentración de proteína a la que la mitad del ADN se une a la proteína es la inversa de K B. Puedes mostrarlo por ti mismo sustituyendo 0.5 por [DP]/[D] tot en la ecuación. En este punto, [P] = 1/K B.
2. Problemas con el enfoque clásico.
En este enfoque clásico, se diseñaron experimentos de tal manera que
o se podría suponer que una o más concentraciones no cambian, y
o las observaciones fueron manipuladas matemáticamente (transformadas) a una ecuación lineal para que uno pudiera
+ trazar los datos transformados,
+ decidir dónde trazar una línea recta, y
+ utilizar la pendiente e intercepciones para estimar los parámetros en cuestión. (Parcelas Scatchard, parcelas Lineweaver-Burke, etc.).
* Dos problemas están asociados con la técnica más antigua
o Decidir dónde dibujar la línea recta es una decisión arbitraria para cada persona que realiza el análisis (y usar una regresión lineal para encontrar la línea de “mejor ajuste” no está justificado, ya que dos de los supuestos sobre sus datos que se necesitan para justificar tal regresión no son ciertos)
o No hay una estimación precisa del error en la estimación del valor del parámetro
3. Estas limitaciones han sido superadas en los últimos 5 años, ayudados por el advenimiento de técnicas de ADN recombinante que permiten la producción de grandes cantidades de las proteínas que se analizan, y la disponibilidad de potentes microcomputadoras que pueden llevar a cabo la gran cantidad de cómputos requeridos para no lineal, análisis de regresión de mínimos cuadrados (NLLS).
a. Podemos modelar reacciones de unión mediante
- tabular los diferentes estados que existen en un sistema,
- asociar cada estado con una probabilidad fraccionaria basada en la función de partición de Boltzmann y la energía libre de Gibb para ese estado (DG),
- y determinar la probabilidad de cualquier medición observada por la relación de
- la suma de probabilidades fraccionarias que dan la observación, y
- la suma de las probabilidades fraccionarias de todos los estados posibles.
Donde j es el número de ligandos unidos, la probabilidad fraccionaria de un estado particular viene dada por esta ecuación para fs.

Como ejemplo, considere un sistema de un solo sitio, como un operador que se une a una proteína. Hay dos estados, el estado 0 sin proteína unida al operador y el estado 1 con una proteína unida. Así se puede escribir la ecuación para f 0 y para f 1.
Si ampliamos las probabilidades fraccionarias para cada una de estas ecuaciones de ocupación fraccional, derivamos ecuaciones que relacionan la ocupación fraccionaria![]() ,, con una función de las energías libres de Gibb para unión (DG), concentración de proteína ([P2]) y estequiometría compleja (j).
,, con una función de las energías libres de Gibb para unión (DG), concentración de proteína ([P2]) y estequiometría compleja (j).
Para un sistema de sitio único, tenemos las siguientes ecuaciones:


Dado que la energía libre de Gibb también está relacionada con la constante de equilibrio para las reacciones:
\[\Delta G= -RT\ln (K_{eq})\]
estas energías libres pueden ser re-fundidas como constantes de equilibrio, de la siguiente manera.

Una presentación más completa de este método, incluyendo un tratamiento de múltiples sitios de unión, se puede obtener en el sitio web de BMB Courses (www.bmb.psu.edu/courses/default.htm) haciendo clic en BMB400 “Nixon Lectures”.
b. Análisis de los datos
Después de recolectar los datos vinculantes, estamos en condiciones de analizar los datos observados para averiguar qué valores para DG o Kb hacen que la función prediga mejor las observaciones. Los estadísticos han desarrollado la Teoría de Máxima Verosimilitud para permitir el uso de los datos para encontrar, para cada parámetro, el valor que es más probable que sea correcto. Para los datos bioquímicos el enfoque más apropiado (la mayor parte del tiempo) es la regresión global, no lineal, de mínimos cuadrados (NLLS).
• Afortunadamente, las computadoras de escritorio son ahora lo suficientemente potentes como para hacer estos cálculos en pocos minutos, para un experimento, o incluso para muchos experimentos combinados en un análisis global. Este método tiene varias ventajas. Te da:
o las mismas estimaciones de parámetros, no importa qué programa o método utilice usted o alguien más, siempre que el programa esté escrito correctamente y se utilice correctamente.
o estimaciones mucho más rigurosas de error.
Este último punto merece la pena destacar:
• ¿no es cierto que $100 (menos $50) es mucho menos atractivo como tarifa por tu tiempo que $100 (menos $0.01)? Lo mismo puede ser cierto para estimaciones de energías libres vinculantes, o constantes de equilibrio.
• Además, cuando se requieren varios experimentos para estimar un parámetro, el error en cada experimento debe incluirse en la estimación del parámetro. Sin un análisis global que determine un error de conglomerado, no es posible llevar con cuidado el error de un experimento al análisis de datos de otros adicionales.
c. Este análisis produce una gráfica de la varianza de ajuste, o error, en un amplio rango de valores posibles para el parámetro que se está midiendo, como el DG para la vinculación. El valor D G con el menor error es el valor más preciso.
Un ejemplo de este análisis se muestra en la Figura 4.2.6. Los datos brutos mostrados en la Figura 4.2.2 (panel izquierdo) produjeron las curvas de unión mostradas en el panel derecho de esa figura. Estos datos se sometieron luego a análisis no lineales de mínimos cuadrados. Los errores (o varianza de ajuste) para cada posible valor de DG se representan en la Figura 4.2.6. Por ejemplo, tenga en cuenta que la varianza más baja de ajuste para DG 1 es de aproximadamente —9.5 kcal/mol.
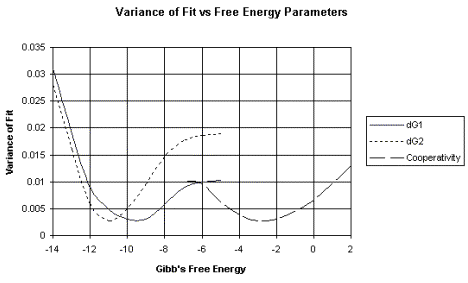
dG1 = DX1 = Energía libre de Gibb para la unión al primer sitio de un sistema de dos sitios.
dG2 = DG2 = Energía libre de Gibb para la unión al segundo sitio de un sistema de dos sitios.
También se representa la varianza de ajuste para la DG para la cooperatividad entre las proteínas unidas en los dos sitios.
Estos datos fueron amablemente proporcionados por la Dra. Tracy Nixon.
Como se indicó anteriormente, una vez que se dispone de un valor para DG, se puede calcular Keq a partir de
DG = -RT ln (Keq)
Figura 4.2.7.
Algunas referencias clave para NLLS:
Senear y Bolen, 1992, Métodos Enzymol. 210:463
Koblan et al, 1992, Methods Enzymol. 210:405.
Senear et al 1991, J. Biol. Chem. 266:13661
E. Insights sobre el mecanismo de regulación lac mediante la medición de constantes de unión
Habiendo pasado por análisis de mínimos cuadrados clásicos y no lineales para medir las constantes de unión, veamos un ejemplo de cómo se utilizan estas mediciones para comprender mejor el mecanismo de regulación génica. Sabemos que la transcripción del operón lac se incrementa en presencia del inductor, pero ¿cómo ocurre esto? Se podría enumerar una serie de posibilidades, cada una con diferentes predicciones sobre cómo el inductor puede afectar la constante de unión del represor para operador, K B.
- ¿El inductor cambia la conformación del represor lac para que ahora active la transcripción? Esto podría ocurrir sin ningún efecto sobre K B.
- ¿El inductor hace que el represor se disocie del ADN del operador y permanezca libre en solución? Esto predice una disminución de K B para ADN específico, pero no unión a ADN inespecífico.
- ¿El inductor hace que el represor se disocie del operador y se redistribuya a sitios inespecíficos en el ADN? Esto predice una disminución de K B para ADN específico, pero propone que la mayor parte del represor está unido a sitios no operarios.
La medición de las constantes de equilibrio para la unión del represor lac al operador y al ADN inespecífico, en ausencia y presencia del inductor, muestra que la posibilidad c anterior es correcta. Esta sección del capítulo explora este resultado en detalle.
En ausencia de inductor, el represor, o R4, se unirá a sitios específicos (en este caso el operador) con alta afinidad y a sitios inespecíficos (otras secuencias de ADN) con menor afinidad (Figura 4.2.8). Esto se establece cuantitativamente en los siguientes valores para la constante de asociación de equilibrio. Cualquiera de las constantes de equilibrio se puede abreviar Keq o KB. Utilizaremos el término KS para referirnos a KB en sitios específicos y KNS para el KB en sitios no específicos.
\[KS = 2 \times 10^{13} M-1 KNS = 2 \times 10^6 M-1 \]
[Una presentación detallada de algunos datos representativos y cómo utilizarlos para determinar estas constantes vinculantes para el represor lac se encuentra en el Apéndice A al final de este capítulo. Este Apéndice pasa por el enfoque clásico para medir las constantes de unión.]
3. La constante de unión del represor lac a su operador cambia en presencia de inductor. (Figura 4.2.8)
La unión del inductor al represor disminuye la afinidad del represor por el operador 1000 veces, pero no afecta la afinidad del represor por sitios inespecíficos.
Para R 4 con inductor:
K S = 2 x 1010 M -1 K NS = 2 x 106 M -1
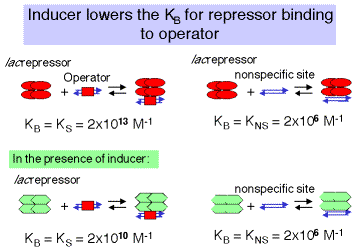
4. La diferencia de afinidad por sitios específicos versus no específicos puede describirse por el parámetro de especificidad, que es la relación entre la constante de equilibrio para la unión específica y la constante de equilibrio para la unión inespecífica.
Especificidad =![]() en ausencia de inductor
en ausencia de inductor
![]() en presencia de inductor
en presencia de inductor
Obsérvese que en presencia del inductor, la especificidad con la que el represor lac se une al ADN disminuye 1000 veces.
Aunque el represor todavía tiene una mayor afinidad por ADN específico en presencia del inductor, hay tantos sitios inespecíficos en el genoma que el represor permanece unido a estos sitios inespecíficos en lugar de encontrar al operador. De ahí que en presencia del inductor, el operador está en gran parte desocupado por el represor, y el operón se transcribe activamente.
La regulación del operón lac a través de la redistribución del represor a sitios inespecíficos del genoma se cubre con más detalle en las dos secciones siguientes. Muestran el efecto de tener un gran número de sitios inespecíficos de baja afinidad compitiendo con un único sitio de alta afinidad por un pequeño número de moléculas represoras.
5.Distribución del represor entre el operador y los sitios no específicos
Aunque el represor tiene una afinidad mucho mayor por el operador que por sitios no específicos, hay muchos más sitios inespecíficos (4.6 x 106, ya que esencialmente cada nucleótido en el genoma de E. coli es el comienzo de un sitio de unión inespecífico) que sitios específicos (un operador por genoma) que prácticamente todo el represor está unido al ADN, aunque solo estén presentes sitios inespecíficos.
- Utilizamos las constantes de unión anteriores, y las acoplamos con un cálculo de que la concentración de represor (10 moléculas por célula) es de 1.7 x 10-8 M y la concentración de sitios inespecíficos (4.6 x 106 por célula) es 7.64 x 10-3 M. Estos valores para [R4] y [DNS] son esencialmente constantes. Con esta información, podemos calcular que la relación de represor libre a la ligada a sitios inespecíficos es menor que 1 x 10-4 (es aproximadamente 6.6 x 10-5), como se muestra en el cuadro de abajo. Por lo tanto, solo aproximadamente 1 de cada 15 mil moléculas represivas no se une al ADN.
- Este análisis muestra que el represor lac se divide entre sitios inespecíficos y el operador. Cuando no está ligado al operador, se une en otra parte a cualquiera de los cerca de 4.6 millones de sitios en el genoma. Casi ninguno de los represores está unido al ADN en la célula.
- El recuadro 2 (abajo) pasa por estos cálculos con más detalle.
Recuadro 2. Efectivamente, todas las proteínas represivas están unidas al ADN.
![]()
![]()
![]()
6. Regulación del operón lac a través de la redistribución del represor a sitios inespecíficos del genoma
a. La alta especificidad del represor para el operador significa que en ausencia de inductor, el operador está unido por el represor prácticamente todo el tiempo. Esto es cierto a pesar del enorme exceso de sitios de unión inespecíficos.
b. El parámetro de especificidad descrito anteriormente (Ks/Kns) permite evaluar los equilibrios simultáneos (represor para operador y represor para sitios inespecíficos en el ADN). Queremos calcular la relación entre los operadores vinculados al represor y los operadores libres. Los valores para KS, KNS y [DNS] ya son conocidos, y la concentración de represor no unido al ADN es insignificante.
Caja 3. El parámetro de especificidad se relaciona con la relación de sitios operarios unidos a libres.
Especificidad =
relación de Límite: Libre
sitios del operador
Ahora necesitamos un valor para [R4DNS]. Esto se obtiene al darse cuenta de que bajo condiciones que saturan sitios específicos, la concentración de represor unido a sitios inespecíficos se aproxima estrechamente por [represor] total - [operador], o [R 4] total - [D s] total en las ecuaciones del Recuadro 4.
Casilla 4.
![]()
[R 4] libre es despreciable (ver arriba).
En condiciones que saturan sitios específicos,
![]()
Así [R 4 D NS] = [R 4] total - [D s] total

c. Después de hacer estas suposiciones simplificadoras, ahora tenemos un valor para cada variable y constante en la ecuación, excepto la relación de sitios de operadores límites:libres. Así podemos calcular la relación deseada.
Caja 5. Ecuación que relaciona la especificidad con la relación de operador unido a libre y un conjunto de constantes.
Especificidad =![]()
ya quiero constantes
medido determinar
d. Ahora que tenemos la ecuación en el Recuadro 5, podemos calcular la relación de operador libre a operador enlazado por represor que se puede calcular en ausencia y presencia de inductor.
(1) En ausencia de inductor:
Especificidad =![]()

es decir, la relación entre operadores libres y operadores vinculados por represor es de 0.05. R 4 está ligado al operador ~ 95% del tiempo. Por lo tanto, el operón no se expresa.
(2) En presencia de inductor:
Especificidad =![]()
![]()
es decir, en presencia de inductor, solo alrededor del 2% de los operadores están vinculados por represor, o R 4 está ligado al operador ~ 2% del tiempo. Así se expresa el operón.
En resumen, estos cálculos muestran que en ausencia de inductor, 95% de los operadores están ocupados (o está obligado por R 4 95% del tiempo). En presencia de inductor, el represor se redistribuye a sitios inespecíficos en el ADN, dejando solo 2% de los operadores unidos por R4. Así, el operón se expresa en la mayoría de las células.
Un ejemplo adicional del uso de las constantes de unión medidas y del parámetro de especificidad se encuentra en el Apéndice B al final de este capítulo. En este ejemplo se exploran los efectos de los mutantes operarios.
F. Mecanismo de represión e inducción para el operón lac
1. Efecto del represor lac sobre la capacidad de la ARN polimerasa para unirse al promotor
El análisis de la sección anterior mostró cómo el inductor afecta la partición del represor entre sitios específicos e inespecíficos. Ahora examinemos el efecto que el represor unido al operador tiene sobre la función de la polimerasa en el promotor
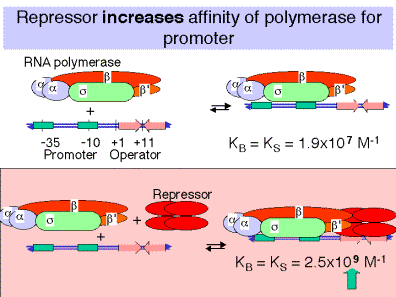
a. ¡La unión del represor al operador en realidad aumenta la afinidad de la ARN polimerasa por el promotor!
Considera el siguiente equilibrio:
RNA polimerasa+ promotor RNA polimerasa-promotor (complejo cerrado)
En ausencia de represor en el operador, la afinidad de la ARN polimerasa por el promotor lac es
KB = 1.9 x 107 M-1
En presencia de represor en el operador, la afinidad es
KB = 2.5 x 109 M-1
b.Represor unido al operador aumenta la afinidad de la ARN polimerasa por el promotor lac aproximadamente 100 veces, por lo que el complejo cerrado se forma mucho más fácilmente. El represor esencialmente mantiene la ARN polimerasa almacenada en el promotor, pero la transcripción no se inicia.
c.Tras la unión del inductor al represor, el represor se disocia y el complejo ARN polimerasa-promotor puede desplazarse al complejo abierto e iniciar la transcripción, activando así el operón.
d.Así, el efecto del represor unido al operador no es sobre Kb para la interacción polimerasa-promotor, sino sobre kf para la conversión de complejo cerrado a abierto.
G. Las mediciones cinéticas de la reacción de iniciación abortiva permiten calcular kf.
1. Ensayo de transcripción abortiva
El complejo de transcripción inicial (ITC) que existe después de la formación del complejo abierto frecuentemente no logra transformarse en el complejo de elongación inicial (IEC). Se libera el producto de ARN, y el sistema se inicia de nuevo. La velocidad a la que se acumulan los transcritos abortados puede proporcionar una medida de la fuerza del promotor, y se han ideado experimentos para usar dicho ensayo para estimar KB para la unión de polimerasa a la región promotora, y kf para isomerización de forma de complejo cerrado a abierto. La polimerasa, el ADN promotor y los nucleótidos se mezclan de tal manera que un fosfato radiomarcado se introducirá en transcritos que se hacen y se abortan. La cantidad de radiactividad en los transcritos cortos se cuenta luego en función del tiempo.
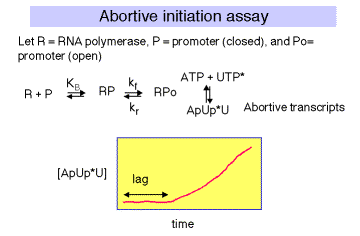
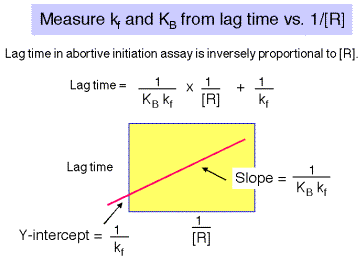
Existe un retraso entre la mezcla de reactivos y la tasa óptima de producción de transcritos abortivos. La duración de este rezago es inversamente proporcional al [RNAP]. Una gráfica de tiempo de retardo vs 1/ [RNAP] da una gráfica de línea recta, con pendiente igual a 1/ [KB 'kf] e intercepción y de 1/kf.
Figura 4.2.11.
H. Activación de la transcripción por la proteína CAP de E. coli
1. La activación de la transcripción por la proteína CAP de E. coli ilustra varios principios reguladores generales.
Nos centraremos en el punto de que en diferentes contextos (diferentes promotores), una sola proteína puede interactuar directamente con RNAP a través de al menos 2 superficies de contacto distintas. Dependiendo del contexto, CAP puede afectar KB o kf para las interacciones ARN polimerasa-promotor.
Una discusión adicional sobre la capacidad de CAP para afectar la arquitectura de un complejo proteína-ADN que contiene contactos precisos entre RNAP y una proteína reguladora adicional (mALT), al doblar el ADN, se encuentra en el sitio web BMB400, bajo “Nixon Lectures”. Este último punto no se abordará en detalle aquí.
2. a Subunidad de la ARN polimerasa
a. Recordemos de la tercera parte que la subunidad a de la ARN polimerasa tiene dos dominios separados. El dominio amino terminal (AntD) es esencial para la dimerización y ensamblaje de la polimerasa, y el dominio carboxi terminal (ActD) es necesario para unirse al ADN y para comunicarse con muchos, pero no todos, factores de transcripción.
La mayoría de la ARN polimerasa (~ 60%) está asociada con genes de ARNr o ARNt. Esto se logra mediante una secuencia especial aguas arriba de los elementos promotores (es decir, las cajas —35 y —10), llamada elemento UP (-57) 5'-AAAATTATTT-3' (-47), que se une a dímeros a2, y aumenta la ocupación por la polimerasa en ~10 veces.
Figura 4.2.12.
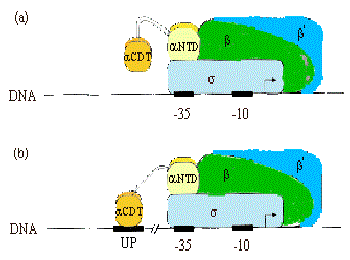
b. Gran parte de la comunicación entre los activadores y la ARN polimerasa de E. coli está mediada entre el CTD de a y estos factores.
(véase Ebright y Busby, 1995, Curr. Opinión en Gen. & Dev. 5:197-203)
Figura 4.2.13.
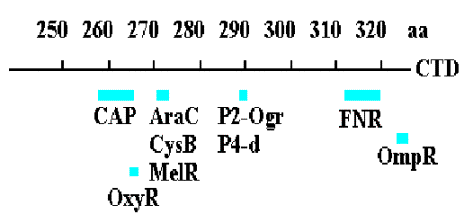
3. Resumen y distinciones entre Cap en Clase I y Cap en Promotores Clase II
(Para opiniones ver Mol Micro 23:853-859 y Cur. Opin. Genet. Dev. 5:197-203).
Los promotores de clase I tienen sitios de unión a CAP centrados en -62, -83 o -93.
En los promotores de clase II, se centra en -42 y se solapa con el determinante -35 del promotor.
Figura 4.2.14. Unión de CAP a promotores de clase I y clase II.
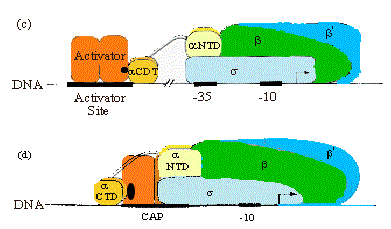
4. CAP tiene al menos dos regiones de activación (AR):
• AR1 (residuos 156-164)
En los promotores de clase I, AR1 en la subunidad aguas abajo de CAP “ve” los residuos 258-265 de CTD de a. Esta interacción aumenta KB para la unión de la polimerasa al promotor.
En los promotores de clase II, CAP desplaza el ActD (KB decreciente), lo que se supera al aumentar KB a través de la interacción subunidad ar1-actD aguas arriba
• AR2 (residuos 19, 21, 96, 101)
En los promotores de clase II, la subunidad aguas abajo “ve” los residuos AntD 162-165, aumentando kf para isomerización de complejos cerrados a abiertos.
Figura 4.2.15. Regiones de activación en CAP
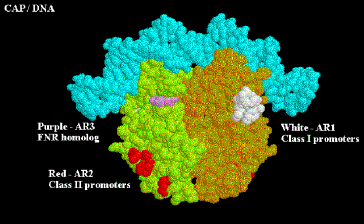
Tanto en los promotores de clase I como de clase II, CAP AR1 interactúa con el CTD de a. Está claro que para los promotores de clase I, los residuos 258-265 de la subunidad a son la diana de AR1 de CAP; no está claro si estos son los mismos residuos necesarios para la interacción en promotores de clase II. En los promotores de clase I, esta interacción proporciona una activación directa “verdadera”: la interacción es entre la subunidad corriente abajo de CAP, y parece que solo se usa para aumentar KB para la unión de la ARN polimerasa a la región promotora (tal vez sustituyendo la falta de una secuencia UP). En los promotores de clase II, AR1 en la subunidad aguas arriba entra en contacto con la subunidad alfa, pero no parece causar estimulación directa de la transcripción. En cambio, supera la inhibición de la polimerasa que se supone que surge de la CAP desplazando a la subunidad alfa de su posición preferida cerca de -45. Esto se evidencia por las siguientes observaciones:
• ActD se une a la región -40 a -55 en promotores de clase II en ausencia de CAP, pero se une a la región -58 a -74 en su presencia
• Los mutantes AR1 en CAP disminuyen KB para ARN polimerasa en promotores de clase II, pero no afectan a kf.
• La eliminación de un CDT elimina la necesidad de CAP AR1 en promotores de clase II, y no tiene ningún efecto negativo.
• En contraste, la eliminación de la CDT impide la activación por CAP en promotores de clase I.
Además de superar una disminución de KB por AR1, en clase II los promotores CAP también ejerce una activación “directa”. Esto ocurre entre los residuos de CAP 19, 21, 96 y 101 (AR2) en la subunidad aguas abajo de CAP, y los residuos 162-165 de la subunidad a NTD. Esta interacción aumenta el kf y no afecta a KB. La región 162-165 se encuentra entre las regiones 30-55/65-75 y 175-185/195-210 que son esenciales para el contacto con las subunidades b y b' de la polimerasa, respectivamente. No se necesita AR2 para que CAP trabaje en promotores de clase I.
Apéndice A para el Capítulo 17 (Cuarta Parte., fracción II)
Medición de constantes de equilibrio para la unión del represor lac a sitios específicos e inespecíficos en ADN
R 4 = Represor
D S = sitio de ADN específico, operador
D NS = sitio de ADN no específico, todos los demás sitios en el genoma
R4 + DS R4DS R4 + DNS R4DNS
![]()
![]()
|
|

![]()
![]()
|
|

![]()
![]()
El represor lac se unirá a su sitio específico, el operador, con muy alta afinidad,
Keq = KS = 2 x 1013 M-1, donde Ks es la constante de asociación de equilibrio para la unión a un sitio específico
y se unirá a otras secuencias de ADN, o sitios inespecíficos, con una mayor afinidad.
Keq = KNS = 2 x 106 M-1, donde Kns es la constante de asociación de equilibrio para la unión a un sitio inespecífico.
Mediciones en el laboratorio:
Dado que puede ser difícil medir la cantidad de sonda unida o libre a concentraciones muy bajas, es más confiable medir la fracción de sonda unida en función de [R4]. La fracción de sonda unida es
=.
Al sustituir [Ds] = [Ds] total- [R4Ds] en la ecuación para Ks, puede derivar la siguiente relación entre la fracción de sonda unida por represor y la concentración del represor:
=
{Dado que el [R4] suele ser mucho mayor que el [Ds] total en estos ensayos, el
[R4] free >> [R4Ds], y [R4] está bien aproximado por [R4] total.}
Esta ecuación tiene la forma de la clásica ecuación de Michaelis-Menten para la cinética enzimática en estado estacionario, y también es útil en el análisis de muchos ensayos de unión. Una vez que se grafica contra [R4], se puede hacer un ajuste de curva para derivar un valor para Ks. También se puede obtener un valor para Ks midiendo el [R4] al que se une la mitad de la sonda. En este punto, [R4] =. {Esto se puede ver simplemente sustituyendo = 0.5 en la ecuación anterior. El álgebra es exactamente el mismo que se hace para la determinación de Km por el análisis de Michaelis-Menten.}
Apéndice B. Uso de constantes de unión y ecuaciones que relacionan el parámetro de especificidad con la relación de sitios operarios unidos a libres para estudiar los efectos de los mutantes operarios.
Las mismas ecuaciones utilizadas en la sección E de este capítulo también pueden utilizarse para examinar los efectos de los mutantes operadores. El siguiente análisis muestra que una mutación que disminuye la afinidad del operador 20 veces por el represor dará como resultado que aproximadamente la mitad de los operadores estén libres de represor (o el operón se exprese aproximadamente la mitad del tiempo).

Especificidad =![]()
![]()
![]()
Esto dice que el operador está esencialmente distribuido equitativamente entre la forma encuadernada y la forma libre.

El 50% de los operadores no están ocupados por represor, por lo que solo se expresará aproximadamente la mitad de los operones (en una población de bacterias), o cualquier operón en particular se expresará aproximadamente la mitad del tiempo.