
2.3: Instrumentos de Microscopía
- Page ID
- 54803

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Objetivos de aprendizaje
- Identificar y describir las partes de un microscopio de campo claro
- Calcular el aumento total para un microscopio compuesto
- Describir las características distintivas y los usos típicos de varios tipos de microscopios de luz, microscopios electrónicos y microscopios de sonda de barrido
Los primeros pioneros de la microscopía abrieron una ventana al mundo invisible de los microorganismos. Pero la microscopía siguió avanzando en los siglos que siguieron. En 1830, Joseph Jackson Lister creó un microscopio óptico esencialmente moderno. El siglo XX vio el desarrollo de microscopios que aprovecharon la luz no visible, como la microscopía de fluorescencia, que utiliza una fuente de luz ultravioleta, y la microscopía electrónica, que utiliza haces de electrones de longitud de onda corta. Estos avances condujeron a importantes mejoras en la ampliación, resolución y contraste. En comparación, los microscopios relativamente rudimentarios de van Leeuwenhoek y sus contemporáneos eran mucho menos poderosos que incluso los microscopios más básicos en uso hoy en día. En esta sección, examinaremos la amplia gama de tecnología microscópica moderna y aplicaciones comunes para cada tipo de microscopio.
Microscopía de luz
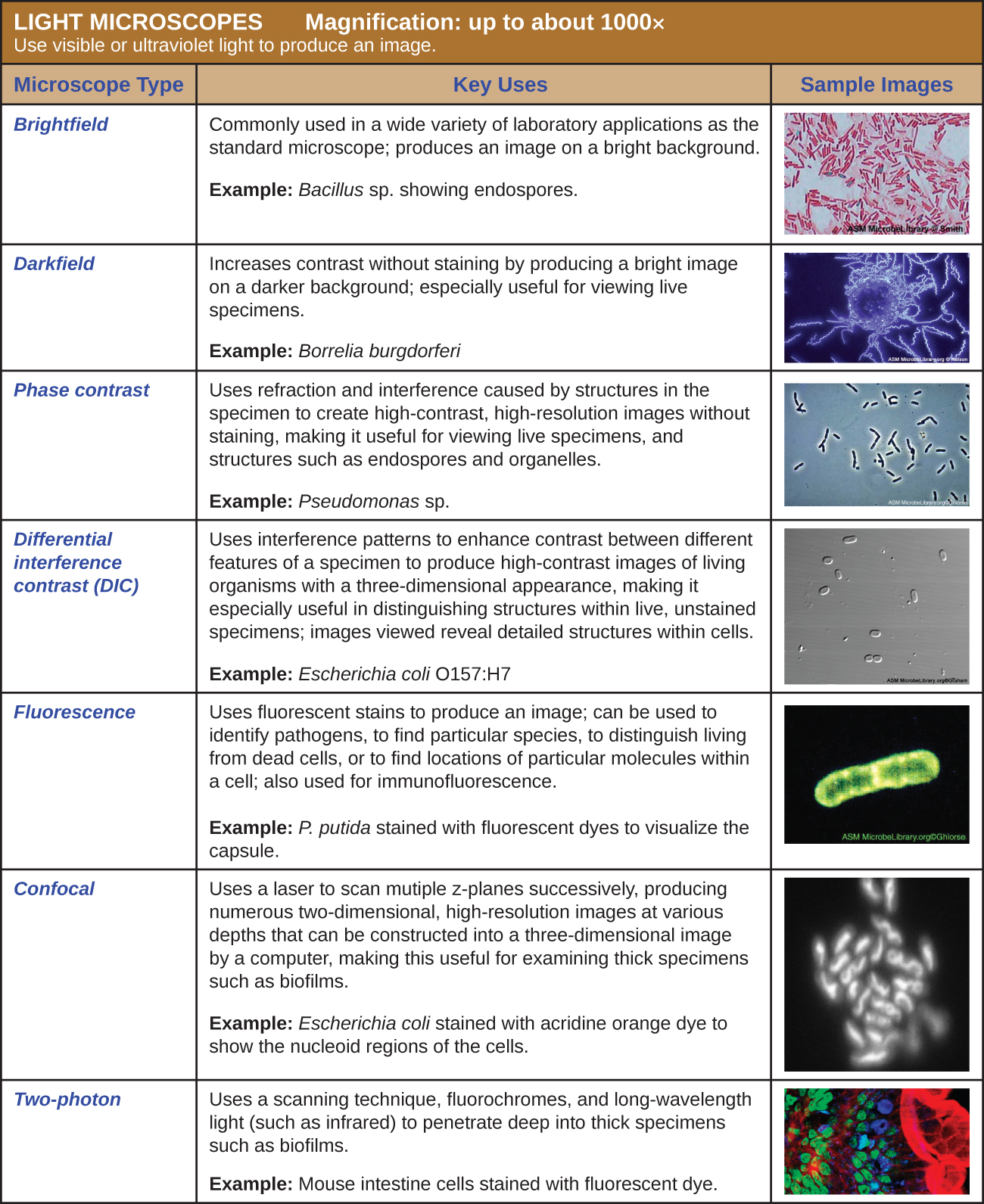
Muchos tipos de microscopios caen dentro de la categoría de microscopios de luz, que utilizan la luz para visualizar imágenes. Ejemplos de microscopios de luz incluyen microscopios de campo claro, microscopios de campo oscuro, microscopios de contraste de fase, microscopios de contraste de interferencia diferencial, microscopios de fluorescencia, microscopios láser de barrido confocal y microscopios de dos fotones. Estos diversos tipos de microscopios de luz se pueden utilizar para complementarse entre sí en diagnósticos e investigación.
Microscopios de campo claro
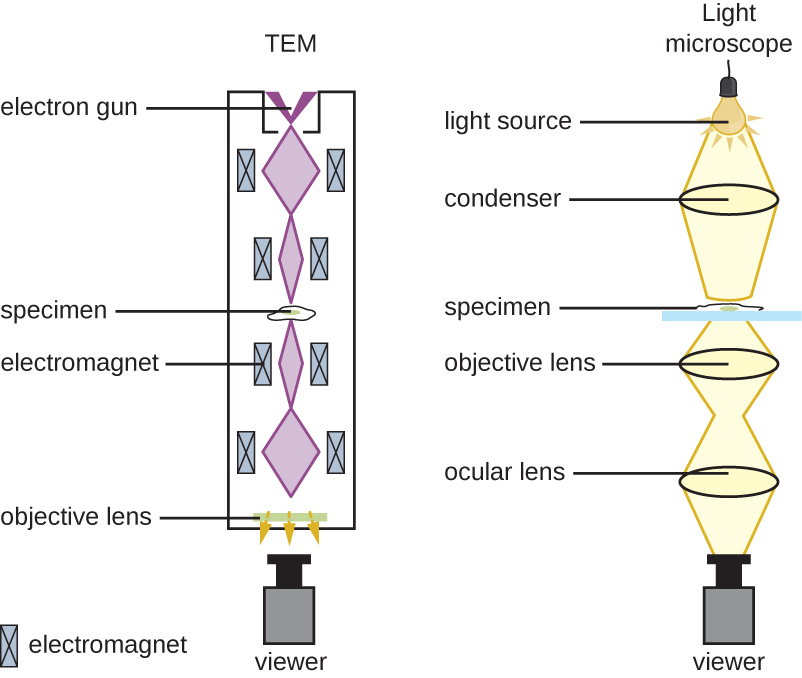
El microscopio de campo claro, quizás el tipo de microscopio más utilizado, es un microscopio compuesto con dos o más lentes que producen una imagen oscura sobre un fondo brillante. Algunos microscopios de campo claro son monoculares (tienen un solo ocular), aunque la mayoría de los microscopios de campo claro más nuevos son binoculares (con dos oculares), como el que se muestra en la Figura\(\PageIndex{1}\); en cualquier caso, cada ocular contiene una lente llamada lente ocular. Las lentes oculares suelen magnificar las imágenes 10 veces (10). En el otro extremo del tubo corporal hay un conjunto de lentes de objetivo en una boquilla giratoria. El aumento de estas lentes de objetivo generalmente varía de 4a 100, con el aumento para cada lente designado en la carcasa metálica de la lente. Los lentes oculares y objetivos trabajan juntos para crear una imagen ampliada. El aumento total es el producto del aumento ocular multiplicado por el aumento objetivo:
\[\text{ocular magnification} \times \text{objective magnification} \nonumber\]
Por ejemplo, si se selecciona una lente\(40 \times\) objetivo y la lente ocular es\(10\times\), el aumento total sería
\[(40×)(10×)=400× \nonumber\]
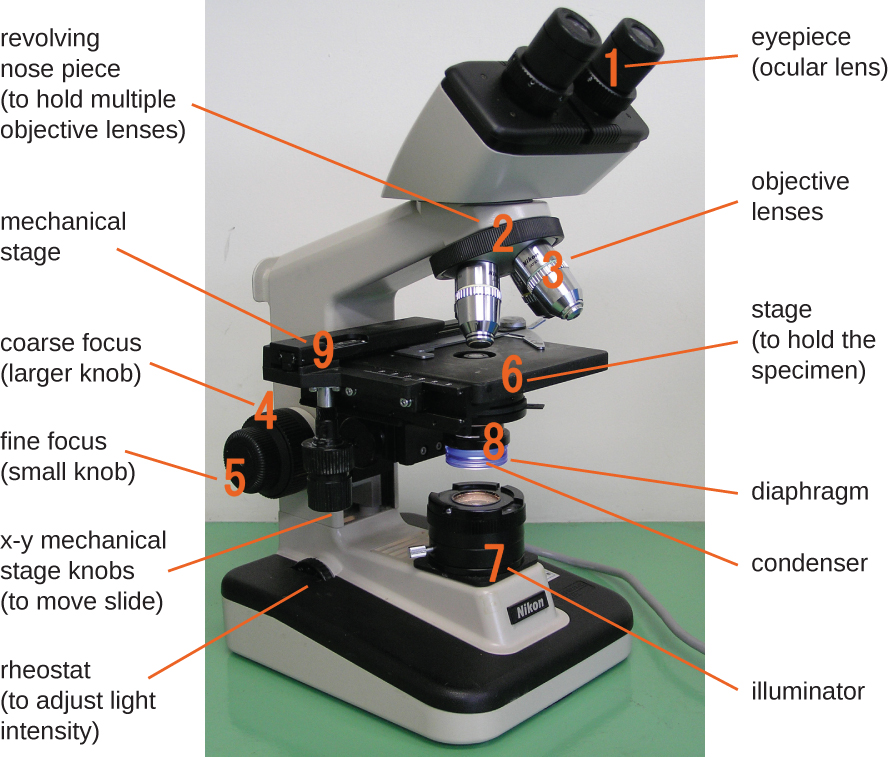
Componentes de un microscopio típico de campo claro.
El elemento que se está viendo se llama espécimen. El espécimen se coloca sobre un portaobjetos de vidrio, que luego se sujeta en su lugar en el escenario (una plataforma) del microscopio. Una vez que el portaobjetos está asegurado, el espécimen en el portaobjetos se coloca sobre la luz usando las perillas mecánicas de escenario x-y. Estas perillas mueven el deslizamiento sobre la superficie del escenario, pero no suben ni bajan el escenario. Una vez que el espécimen está centrado sobre la luz, la posición del escenario puede elevarse o bajarse para enfocar la imagen. La perilla de enfoque grueso se utiliza para movimientos a gran escala con lentes de objetivo de 4y 10; la perilla de enfoque fino se utiliza para movimientos a pequeña escala, especialmente con lentes de objetivo de 40o 100.
Cuando las imágenes se magnifican, se vuelven más tenues porque hay menos luz por unidad de área de imagen. Las imágenes altamente ampliadas producidas por microscopios, por lo tanto, requieren una iluminación intensa. En un microscopio de campo claro, esta luz es proporcionada por un iluminador, que suele ser una bombilla de alta intensidad debajo del escenario. La luz del iluminador pasa hacia arriba a través de la lente condensadora (ubicada debajo del escenario), que enfoca todos los rayos de luz en la muestra para maximizar la iluminación. La posición del condensador se puede optimizar utilizando el botón de enfoque del condensador adjunto; una vez establecida la distancia óptima, el condensador no debe moverse para ajustar el brillo. Si se necesitan niveles de luz menores que los máximos, la cantidad de luz que golpea la muestra se puede ajustar fácilmente abriendo o cerrando un diafragma entre el condensador y la muestra. En algunos casos, el brillo también se puede ajustar utilizando el reóstato, un dimmer switch que controla la intensidad del iluminador.
Un microscopio de campo claro crea una imagen dirigiendo la luz del iluminador hacia el espécimen; esta luz es transmitida, absorbida, reflejada o refractada diferencialmente por diferentes estructuras. Los diferentes colores pueden comportarse de manera diferente ya que interactúan concromóforos (pigmentos que absorben y reflejan longitudes de onda particulares de la luz) en partes del espécimen. A menudo, los cromóforos se agregan artificialmente al espécimen usando manchas, que sirven para aumentar el contraste y la resolución. En general, las estructuras en el espécimen aparecerán más oscuras, en diversos grados, que el fondo brillante, creando imágenes máximamente nítidas a aumentos de hasta aproximadamente 1000. Un aumento adicional crearía una imagen más grande, pero sin mayor resolución. Esto nos permite ver objetos tan pequeños como bacterias, que son visibles a aproximadamente 400más o menos, pero no objetos más pequeños como los virus.
Con aumentos muy altos, la resolución puede verse comprometida cuando la luz pasa a través de la pequeña cantidad de aire entre el espécimen y la lente. Esto se debe a la gran diferencia entre los índices de refracción del aire y el vidrio; el aire dispersa los rayos de luz antes de que puedan ser enfocados por la lente. Para resolver este problema, se puede utilizar una gota de aceite para llenar el espacio entre el espécimen y una lente de inmersión en aceite, una lente especial diseñada para ser utilizada con aceites de inmersión. Dado que el aceite tiene un índice de refracción muy similar al del vidrio, aumenta el ángulo máximo en el que la luz que sale del espécimen puede golpear la lente. Esto aumenta la luz recogida y, así, la resolución de la imagen (Figura\(\PageIndex{2}\)). Una variedad de aceites se pueden utilizar para diferentes tipos de luz.
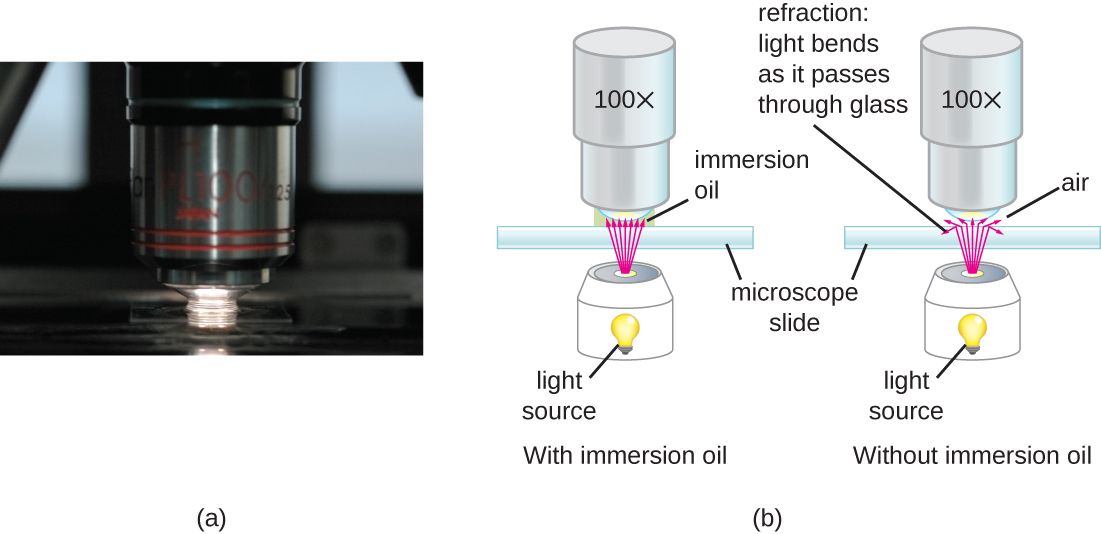
Incluso un microscopio muy potente no puede entregar imágenes de alta resolución si no se limpia y mantiene adecuadamente. Dado que las lentes están cuidadosamente diseñadas y fabricadas para refractar la luz con un alto grado de precisión, incluso una lente ligeramente sucia o rayada refractará la luz de manera no intencionada, degradando la imagen del espécimen. Además, los microscopios son instrumentos bastante delicados, y se debe tener mucho cuidado para evitar dañar partes y superficies. Entre otras cosas, el cuidado adecuado de un microscopio incluye lo siguiente:
- limpiar las lentes con papel para lentes
- no permitir que las lentes entren en contacto con el portaobjetos (por ejemplo, cambiando rápidamente el enfoque)
- proteger la bombilla (si la hay) de roturas
- no empujar un objetivo en una diapositiva
- no usar la perilla de enfoque grueso cuando se usan las lentes de objetivo de 40o mayores
- solo usando aceite de inmersión con un objetivo de aceite especializado, generalmente el objetivo de 100
- aceite de limpieza de lentes de inmersión después de usar el microscopio
- limpiar cualquier aceite transferido accidentalmente de otras lentes
- cubrir el microscopio o colocarlo en un gabinete cuando no esté en uso
Microscopía de campo oscuro
Un microscopio de campo oscuro es un microscopio de campo claro que tiene una modificación pequeña pero significativa en el condensador. Un disco pequeño y opaco (de aproximadamente 1 cm de diámetro) se coloca entre el iluminador y la lente condensadora. Esta parada de luz opaca, como se le llama al disco, bloquea la mayor parte de la luz del iluminador a medida que pasa a través del condensador en su camino hacia la lente objetivo, produciendo un cono hueco de luz que se enfoca en el espécimen. La única luz que alcanza el objetivo es la luz que ha sido refractada o reflejada por estructuras en el espécimen. La imagen resultante suele mostrar objetos brillantes sobre un fondo oscuro (Figura\(\PageIndex{3}\))
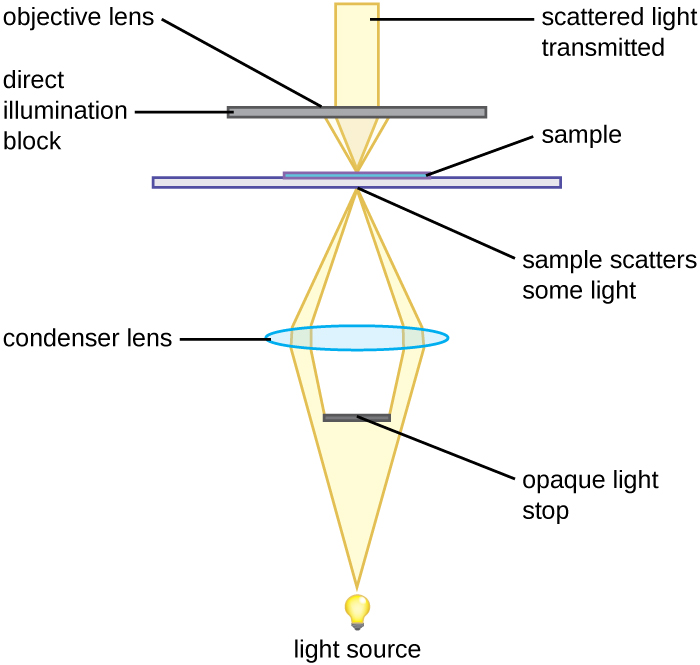
Se utiliza un tope de luz opaco insertado en un microscopio de campo claro para producir una imagen de campo oscuro. La parada de luz bloquea la luz que viaja directamente desde el iluminador hasta la lente del objetivo, permitiendo que solo la luz reflejada o refractada del espécimen llegue al ojo.
La microscopía de campo oscuro a menudo puede crear imágenes de alto contraste y alta resolución de especímenes sin el uso de manchas, lo que es particularmente útil para ver especímenes vivos que podrían morir o verse comprometidos por las manchas. Por ejemplo, las espiroquetas delgadas como Treponema pallidum, el agente causante de la sífilis, se pueden ver mejor usando un microscopio de campo oscuro (Figura\(\PageIndex{4}\)).
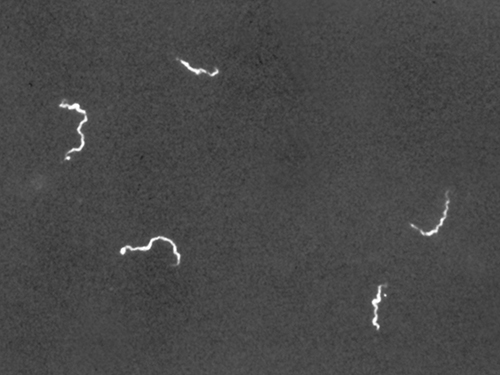
El uso de un microscopio de campo oscuro nos permite ver muestras vivas, sin teñir, de la espiroqueta Treponema pallidum. Similar a un negativo fotográfico, las espiroquetas aparecen brillantes sobre un fondo oscuro. (crédito: Centros para el Control y Prevención de Enfermedades/C.W. Hubbard)
Ejercicio\(\PageIndex{1}\)
Identificar las diferencias clave entre microscopía de campo claro y campo oscuro.
Enfoque Clínico: Parte 2
Las infecciones de heridas como la de Cindy pueden ser causadas por muchos tipos diferentes de bacterias, algunas de las cuales pueden propagarse rápidamente con complicaciones graves. Identificar la causa específica es muy importante para seleccionar un medicamento que pueda matar o detener el crecimiento de la bacteria.
Después de llamar a un médico local sobre el caso de Cindy, la enfermera del campamento envía la muestra de la herida al laboratorio médico más cercano. Desafortunadamente, dado que el campamento se encuentra en una zona remota, el laboratorio más cercano es pequeño y está mal equipado. Un laboratorio más moderno probablemente usaría otros métodos para cultivar, cultivar e identificar las bacterias, pero en este caso, el técnico decide hacer una montura húmeda a partir del espécimen y visualizarla bajo un microscopio de campo claro. En una montura húmeda, se agrega una pequeña gota de agua al portaobjetos, y se coloca un cubreobjetos sobre el espécimen para mantenerlo en su lugar antes de colocarlo debajo de la lente del objetivo.
Bajo el microscopio de campo claro, el técnico apenas puede ver las células bacterianas porque son casi transparentes contra el fondo brillante. Para aumentar el contraste, el técnico inserta un tope de luz opaco sobre el iluminador. La imagen resultante del campo oscuro muestra claramente que las células bacterianas son esféricas y agrupadas en racimos, como uvas.
- ¿Por qué es importante identificar la forma y los patrones de crecimiento de las células en un espécimen?
- ¿Qué otros tipos de microscopía se podrían utilizar de manera efectiva para visualizar este espécimen?
Microscopios de contraste de fase
Los microscopios de contraste de fase utilizan refracción e interferencia causadas por estructuras en una muestra para crear imágenes de alto contraste y alta resolución sin tinción. Es el tipo de microscopio más antiguo y simple que crea una imagen alterando las longitudes de onda de los rayos de luz que pasan a través del espécimen. Para crear trayectorias de longitud de onda alteradas, se utiliza un tope anular en el condensador. El tope anular produce un cono hueco de luz que se enfoca sobre el espécimen antes de alcanzar la lente objetivo. El objetivo contiene una placa de fase que contiene un anillo de fase. Como resultado, la luz que viaja directamente desde el iluminador pasa a través del anillo de fase mientras que la luz refractada o reflejada por la muestra pasa a través de la placa. Esto hace que las ondas que viajan a través del anillo estén aproximadamente la mitad de una longitud de onda fuera de fase con las que pasan a través de la placa. Debido a que las olas tienen picos y valles, pueden sumarse (si están en fase juntas) o cancelarse entre sí (si están fuera de fase). Cuando las longitudes de onda están desfasadas, los valles de onda cancelarán los picos de onda, lo que se denomina interferencia destructiva. Las estructuras que refractan la luz aparecen entonces oscuras contra un fondo brillante de solo luz no refractada. De manera más general, las estructuras que difieren en características como el índice de refracción diferirán en los niveles de oscuridad (Figura\(\PageIndex{5}\)).
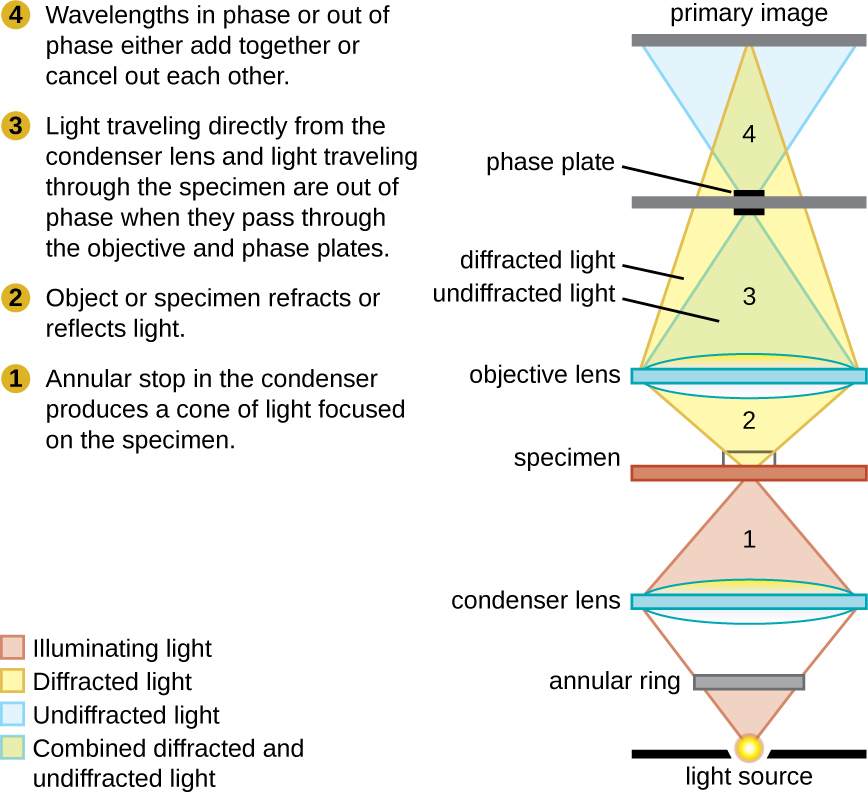
Este diagrama de un microscopio de contraste de fase ilustra las diferencias de fase entre la luz que pasa a través del objeto y el fondo. Estas diferencias se producen al pasar los rayos a través de diferentes partes de una placa de fase. Los rayos de luz se superponen en el plano de la imagen, produciendo contraste debido a su interferencia.
Debido a que aumenta el contraste sin requerir manchas, la microscopía de contraste de fase se utiliza a menudo para observar especímenes vivos. Ciertas estructuras, como los orgánulos en células eucariotas y las endoesporas en células procariotas, están especialmente bien visualizadas con microscopía de contraste de fase (Figura\(\PageIndex{6}\)).
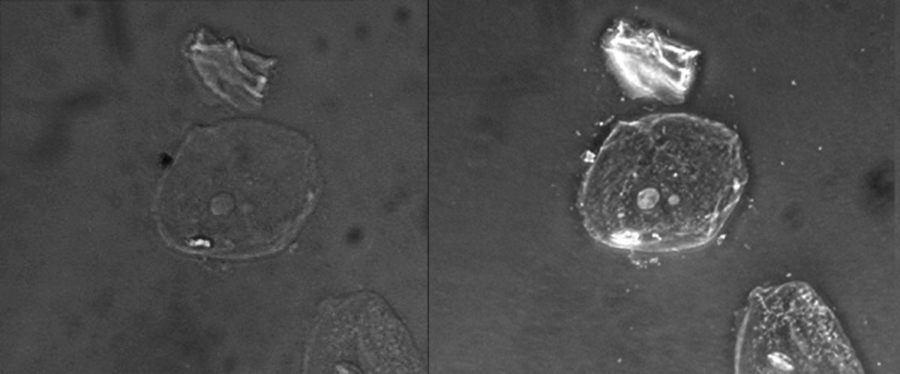
Esta figura compara una imagen de campo claro (izquierda) con una imagen de contraste de fase (derecha) de las mismas células epiteliales escamosas simples sin teñir. Las células están en el centro e inferior derecha de cada fotografía (el elemento irregular sobre las células son restos acelulares). Observe que las células sin teñir en la imagen de campo claro son casi invisibles contra el fondo, mientras que las células en la imagen de contraste de fase parecen brillar contra el fondo, revelando muchos más detalles. (crédito: “Claramente kéfir” /Wikimedia Commons)
Microscopios de contraste de interferencia diferencial
Los microscopios de contraste de interferencia diferencial (DIC) (también conocidos como óptica Nomarski) son similares a los microscopios de contraste de fase en que utilizan patrones de interferencia para mejorar el contraste entre las diferentes características de un espécimen. En un microscopio DIC, se crean dos haces de luz en los que difiere la dirección del movimiento de las ondas (polarización). Una vez que los haces pasan a través del espécimen o del espacio libre de especímenes, se recombinan y los efectos de los especímenes causan diferencias en los patrones de interferencia generados por la combinación de los haces. Esto da como resultado imágenes de alto contraste de organismos vivos con una apariencia tridimensional. Estos microscopios son especialmente útiles para distinguir estructuras dentro de especímenes vivos y sin teñir. (Figura\(\PageIndex{7}\)).

Imagen DIC de Fonsecaea pedrosoi cultivada en agar leoniano modificado. Este hongo causa cromoblastomicosis, una infección crónica de la piel común en climas tropicales y subtropicales.
Ejercicio\(\PageIndex{2}\)
¿Cuáles son algunas ventajas del contraste de fase y la microscopía DIC?
Microscopios de fluorescencia
Un microscopio de fluorescencia utiliza cromóforos fluorescentes llamados fluorocromos, que son capaces de absorber energía de una fuente de luz y luego emitir esta energía como luz visible. Los fluorocromos incluyen sustancias naturalmente fluorescentes (como clorofilas) así como manchas fluorescentes que se agregan al espécimen para crear contraste. Colorantes como rojo Texas y FITC son ejemplos de fluorocromos. Otros ejemplos incluyen los colorantes de ácido nucleico 4',6'-diamidino-2-fenilindol (DAPI) y naranja acridina.
El microscopio transmite una luz de excitación, generalmente una forma de EMR con una longitud de onda corta, como la luz ultravioleta o azul, hacia el espécimen; los cromóforos absorben la luz de excitación y emiten luz visible con longitudes de onda más largas. Luego se filtra la luz de excitación (en parte porque la luz ultravioleta es dañina para los ojos) de manera que solo la luz visible pasa a través del cristalino ocular. Esto produce una imagen del ejemplar en colores brillantes sobre un fondo oscuro.
Los microscopios de fluorescencia son especialmente útiles en microbiología clínica. Se pueden utilizar para identificar patógenos, para encontrar especies particulares dentro de un ambiente, o para encontrar las ubicaciones de moléculas y estructuras particulares dentro de una célula. También se han desarrollado enfoques para distinguir las células vivas de las muertas usando microscopía de fluorescencia basándose en si absorben fluorocromos particulares. En ocasiones, se utilizan múltiples fluorocromos en el mismo espécimen para mostrar diferentes estructuras o características.
Una de las aplicaciones más importantes de la microscopía de fluorescencia es una técnica llamada inmunofluorescencia, que se utiliza para identificar ciertos microbios causantes de enfermedades al observar si los anticuerpos se unen a ellos. (Los anticuerpos son moléculas proteicas producidas por el sistema inmunitario que se adhieren a patógenos específicos para matarlos o inhibirlos). Existen dos enfoques para esta técnica: el ensayo de inmunofluorescencia directa (DFA) y el ensayo de inmunofluorescencia indirecta (IFA). En DFA, los anticuerpos específicos (por ejemplo, aquellos que se dirigen al virus de la rabia) se tiñen con un fluorocromo. Si el espécimen contiene el patógeno diana, se pueden observar los anticuerpos que se unen al patógeno bajo el microscopio fluorescente. Esto se denomina tinción de anticuerpos primarios porque los anticuerpos teñidos se unen directamente al patógeno.
En IFA, los anticuerpos secundarios se tiñen con un fluorocromo en lugar de anticuerpos primarios. Los anticuerpos secundarios no se unen directamente al patógeno, pero sí se unen a anticuerpos primarios. Cuando los anticuerpos primarios no teñidos se unen al patógeno, se puede observar que los anticuerpos secundarios fluorescentes se unen a los anticuerpos primarios. Así, los anticuerpos secundarios se unen indirectamente al patógeno. Dado que múltiples anticuerpos secundarios a menudo pueden unirse a un anticuerpo primario, IFA aumenta el número de anticuerpos fluorescentes unidos al espécimen, lo que facilita la visualización de las características en el espécimen (Figura\(\PageIndex{8}\)).
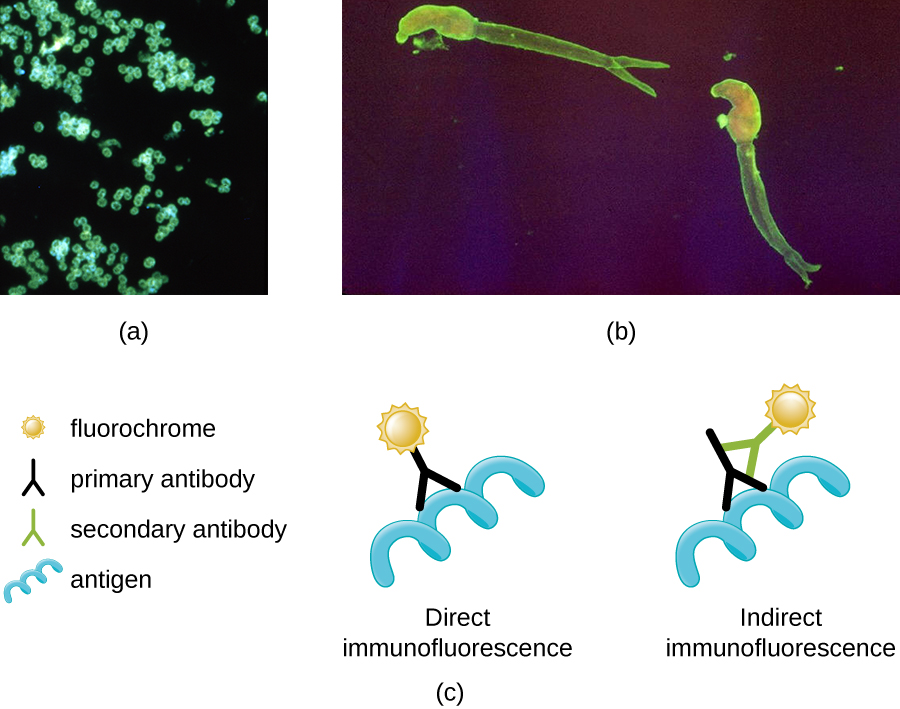
Ejercicio\(\PageIndex{3}\)
¿Por qué se deben usar fluorocromos para examinar una muestra bajo un microscopio de fluorescencia?
Microscopios confocales
Mientras que otras formas de microscopía óptica crean una imagen que se enfoca al máximo a una sola distancia del observador (la profundidad o plano z), un microscopio confocal utiliza un láser para escanear varios planos z sucesivamente. Esto produce numerosas imágenes bidimensionales y de alta resolución a diversas profundidades, que pueden ser construidas en una imagen tridimensional por una computadora. Al igual que con los microscopios de fluorescencia, las manchas fluorescentes se utilizan generalmente para aumentar el contraste y la resolución La claridad de la imagen se ve reforzada aún más por una abertura estrecha que elimina cualquier luz que no sea del plano z. Por lo tanto, los microscopios confocales son muy útiles para examinar especímenes gruesos como biopelículas, los cuales pueden ser examinados vivos y sin fijar (Figura\(\PageIndex{9}\)).
Microscopios de dos fotones
Si bien los microscopios fluorescentes y confocales originales permitieron una mejor visualización de características únicas en especímenes, aún hubo problemas que impidieron una visualización óptima. La sensibilidad efectiva de la microscopía de fluorescencia al ver especímenes gruesos generalmente estuvo limitada por destellos fuera de foco, lo que resultó en una mala resolución. Esta limitación se redujo en gran medida en el microscopio confocal mediante el uso de un estenopeico confocal para rechazar la fluorescencia de fondo fuera de foco con secciones ópticas delgadas (<1 μm) no borrosas. Sin embargo, incluso los microscopios confocales carecían de la resolución necesaria para ver muestras de tejido grueso. Estos problemas se resolvieron con el desarrollo del microscopio de dos fotones, que utiliza una técnica de escaneo, fluorocromos y luz de longitud de onda larga (como la infrarroja) para visualizar especímenes. La baja energía asociada con la luz de longitud de onda larga significa que dos fotones deben golpear una ubicación al mismo tiempo para excitar el fluorocromo. La baja energía de la luz de excitación es menos dañina para las células, y la longitud de onda larga de la luz de excitación penetra más fácilmente en especímenes gruesos. Esto hace que el microscopio de dos fotones sea útil para examinar células vivas dentro de tejidos intactos: cortes de cerebro, embriones, órganos completos e incluso animales enteros.
Actualmente, el uso de microscopios de dos fotones se limita a laboratorios clínicos y de investigación avanzados debido a los altos costos de los instrumentos. Un solo microscopio de dos fotones generalmente cuesta entre 300,000 y 500,000 dólares, y los láseres utilizados para excitar los tintes utilizados en las muestras también son muy caros. Sin embargo, a medida que mejora la tecnología, los microscopios de dos fotones pueden estar más fácilmente disponibles en entornos clínicos.
Ejercicio\(\PageIndex{4}\)
¿Qué tipos de especímenes se examinan mejor mediante microscopía confocal o de dos fotones?
Microscopía Electrónica
La resolución teórica máxima de las imágenes creadas por microscopios de luz está limitada en última instancia por las longitudes de onda de la luz visible. La mayoría de los microscopios de luz solo pueden magnificar 1000, y unos pocos pueden aumentar hasta 1500, pero esto no comienza a acercarse a la potencia de aumento de un microscopio electrónico (EM), que utiliza haces de electrones de longitud de onda corta en lugar de luz para aumentar el aumento y la resolución.
Los electrones, al igual que la radiación electromagnética, pueden comportarse como ondas, pero con longitudes de onda de 0.005 nm, pueden producir una resolución mucho mejor que la luz visible. Un EM puede producir una imagen nítida que se magnifica hasta 100,000. Por lo tanto, los EM pueden resolver estructuras subcelulares así como algunas estructuras moleculares (por ejemplo, cadenas simples de ADN); sin embargo, la microscopía electrónica no se puede usar en material vivo debido a los métodos necesarios para preparar los especímenes.
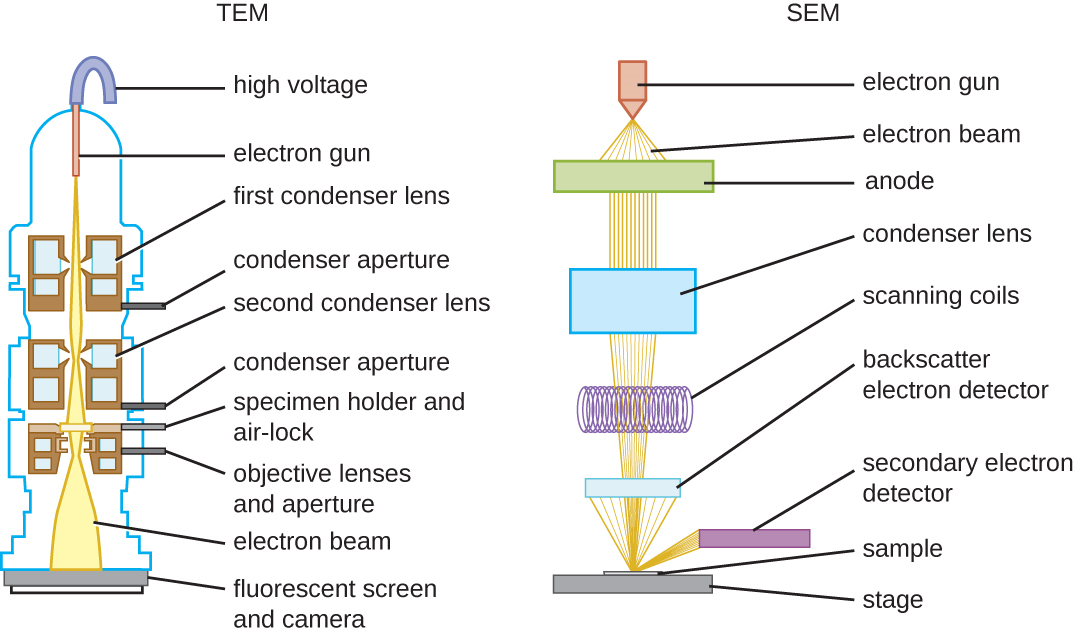
Existen dos tipos básicos de EM: el microscopio electrónico de transmisión (TEM) y el microscopio electrónico de barrido (SEM) (Figura\(\PageIndex{10}\)). El TEM es algo análogo al microscopio de luz de campo claro en términos de su funcionamiento. Sin embargo, utiliza un haz de electrones desde arriba del espécimen que se enfoca usando una lente magnética (en lugar de una lente de vidrio) y se proyecta a través de la muestra sobre un detector. Los electrones pasan a través del espécimen, y luego el detector captura la imagen (Figura\(\PageIndex{11}\)).
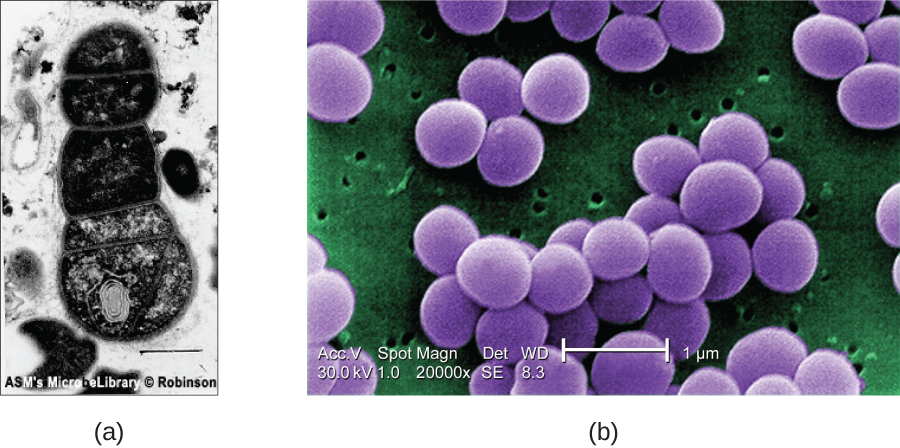
Para que los electrones pasen a través del espécimen en un TEM, el espécimen debe ser extremadamente delgado (20—100 nm de espesor). La imagen se produce debido a la opacidad variable en varias partes del espécimen. Esta opacidad se puede mejorar al teñir el espécimen con materiales como metales pesados, que son densos de electrones. TEM requiere que la viga y el espécimen estén al vacío y que el espécimen sea muy delgado y deshidratado. Los pasos específicos necesarios para preparar un espécimen para su observación bajo un EM se discuten en detalle en la siguiente sección.
Los SEM forman imágenes de superficies de especímenes, generalmente a partir de electrones que son derribados de especímenes por un haz de electrones. Esto puede crear imágenes altamente detalladas con una apariencia tridimensional que se muestran en un monitor (Figura\(\PageIndex{12}\)). Por lo general, los especímenes se secan y preparan con fijadores que reducen los artefactos, como el marchitamiento, que se pueden producir por secado, antes de ser recubiertos por pulverización catódica con una fina capa de metal como el oro. Mientras que la microscopía electrónica de transmisión requiere secciones muy delgadas y permite ver estructuras internas como orgánulos y el interior de membranas, la microscopía electrónica de barrido puede usarse para ver las superficies de objetos más grandes (como un grano de polen) así como las superficies de muestras muy pequeñas ( Figura\(\PageIndex{13}\)). Algunos EM pueden ampliar una imagen hasta 2,000,000. 1
Ejercicio\(\PageIndex{5}\)
- ¿Cuáles son algunas ventajas y desventajas de la microscopía electrónica, a diferencia de la microscopía óptica, para examinar especímenes microbiológicos?
- ¿Qué tipos de especímenes se examinan mejor con TEM? SEM?
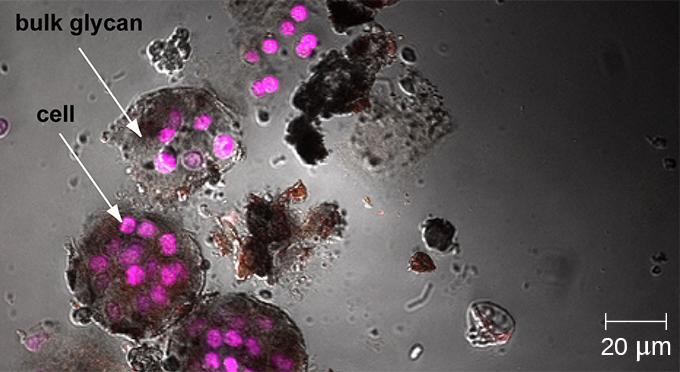
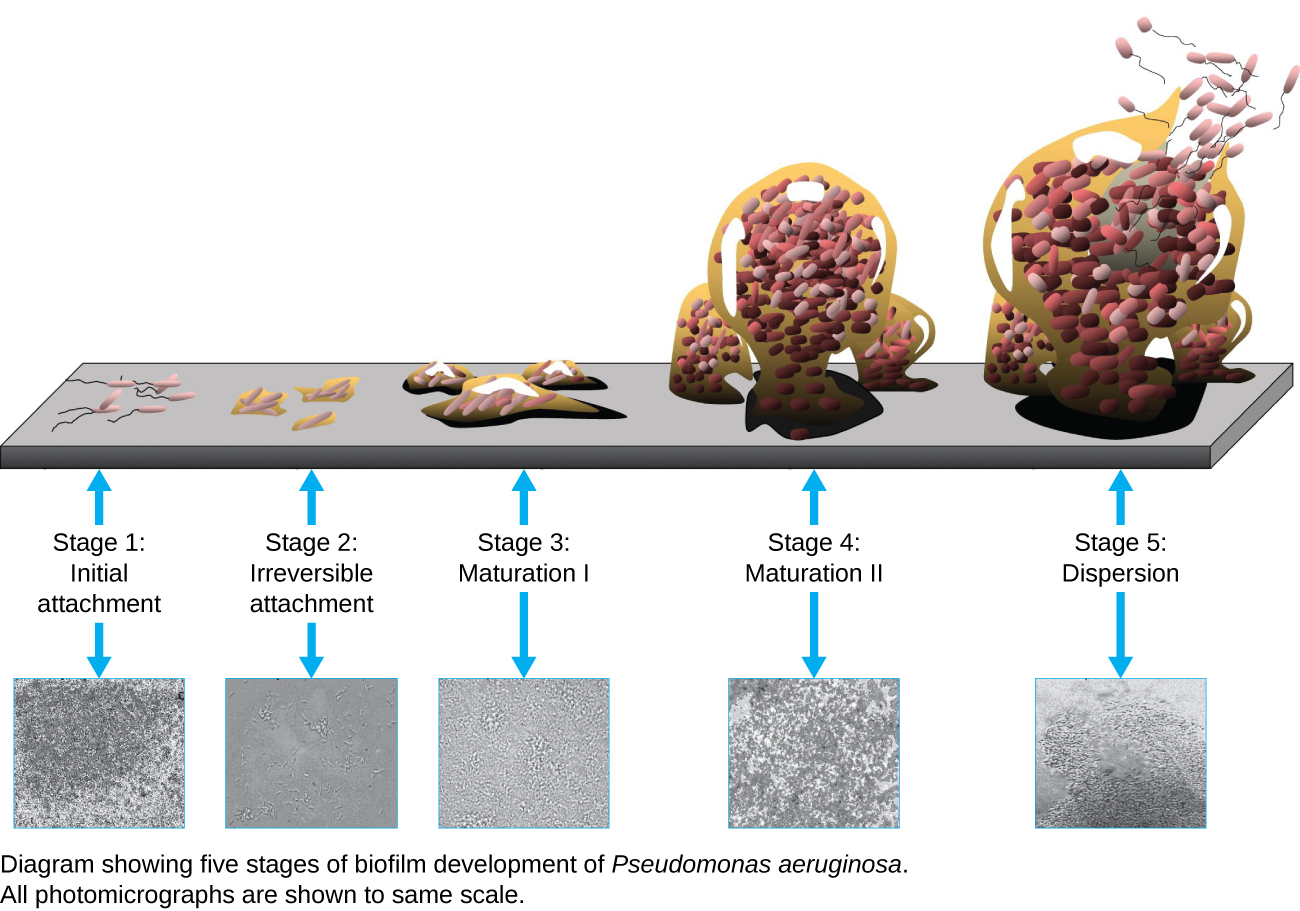
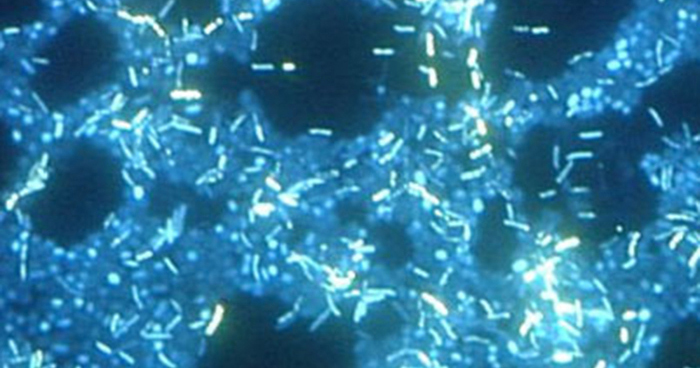
Una biopelícula es una comunidad compleja de una o más especies de microorganismos, que típicamente se forma como un recubrimiento viscoso unido a una superficie debido a la producción de una sustancia extrapolímera (EPS) que se adhiere a una superficie o en la interfaz entre las superficies (por ejemplo, entre el aire y el agua). En la naturaleza, las biopelículas son abundantes y frecuentemente ocupan nichos complejos dentro de los ecosistemas (Figura\(\PageIndex{14}\)). En medicina, las biopelículas pueden cubrir los dispositivos médicos y existir dentro del cuerpo. Debido a que poseen características únicas, como una mayor resistencia contra el sistema inmune y a los medicamentos antimicrobianos, las biopelículas son de particular interés tanto para microbiólogos como para médicos.
Debido a que las biopelículas son gruesas, no se pueden observar muy bien mediante microscopía óptica; cortar una biopelícula para crear un espécimen más delgado podría matar o perturbar a la comunidad microbiana. La microscopía confocal proporciona imágenes más claras de biopelículas porque puede enfocarse en un plano z a la vez y producir una imagen tridimensional de un espécimen grueso. Los colorantes fluorescentes pueden ser útiles para identificar células dentro de la matriz. Adicionalmente, se pueden utilizar técnicas como la inmunofluorescencia y la hibridación fluorescente in situ (FISH), en las que se utilizan sondas fluorescentes para unirse al ADN.
La microscopía electrónica puede ser utilizada para observar biopelículas, pero solo después de deshidratar el espécimen, lo que produce artefactos indeseables y distorsiona el espécimen. Además de estos enfoques, es posible seguir las corrientes de agua a través de las formas (como conos y hongos) de las biopelículas, utilizando video del movimiento de perlas recubiertas fluorescentemente (Figura\(\PageIndex{15}\)).
Microscopía con sonda de barrido
Un microscopio de sonda de barrido no utiliza luz ni electrones, sino sondas muy afiladas que pasan sobre la superficie del espécimen e interactúan con él directamente. Esto produce información que se puede ensamblar en imágenes con aumentos de hasta 100,000,000. Tales ampliaciones grandes se pueden utilizar para observar átomos individuales en superficies. Hasta la fecha, estas técnicas se han utilizado principalmente para la investigación y no para el diagnóstico.
Hay dos tipos de microscopios con sonda de barrido: el microscopio de túnel de barrido (STM) y el microscopio de fuerza atómica (AFM). Un STM utiliza una sonda que se pasa justo por encima de la muestra, ya que una polarización de voltaje constante crea el potencial de una corriente eléctrica entre la sonda y la muestra. Esta corriente se produce a través de un túnel cuántico de electrones entre la sonda y el espécimen, y la intensidad de la corriente depende de la distancia entre la sonda y el espécimen. La sonda se mueve horizontalmente por encima de la superficie y se mide la intensidad de la corriente. La microscopía de túnel de barrido puede mapear efectivamente la estructura de las superficies a una resolución en la que se pueden detectar átomos individuales.
Similar a un STM, los AFM tienen una sonda delgada que se pasa justo por encima del espécimen. Sin embargo, en lugar de medir variaciones en la corriente a una altura constante por encima del espécimen, un AFM establece una corriente constante y mide variaciones en la altura de la punta de la sonda a medida que pasa sobre el espécimen. A medida que la punta de la sonda pasa sobre el espécimen, las fuerzas entre los átomos (fuerzas de van der Waals, fuerzas capilares, unión química, fuerzas electrostáticas y otras) hacen que se mueva hacia arriba y hacia abajo. La deflexión de la punta de la sonda se determina y mide utilizando la ley de elasticidad de Hooke, y esta información se utiliza para construir imágenes de la superficie del espécimen con resolución a nivel atómico (Figura\(\PageIndex{16}\)).

Ejercicio\(\PageIndex{6}\)
- ¿Cuál tiene mayor aumento, un microscopio óptico o un microscopio de sonda de barrido?
- Nombra una ventaja y una limitación de la microscopía con sonda de barrido.


Conceptos clave y resumen
- Numerosos tipos de microscopios utilizan diversas tecnologías para generar micrografías. La mayoría son útiles para un tipo particular de espécimen o aplicación.
- La microscopía óptica utiliza lentes para enfocar la luz en un espécimen para producir una imagen. Los microscopios de luz comúnmente utilizados incluyen microscopios de campo claro, campo oscuro, contraste de fase, contraste de interferencia diferencial, fluorescencia, confocales y microscopios de dos fotones.
- La microscopía electrónica enfoca los electrones en el espécimen usando imanes, produciendo un aumento mucho mayor que la microscopía óptica. El microscopio electrónico de transmisión (TEM) y el microscopio electrónico de barrido (SEM) son dos formas comunes.
- La microscopía de sonda de barrido produce imágenes de aumento aún mayor al medir la retroalimentación de sondas afiladas que interactúan con la muestra. Los microscopios de sonda incluyen el microscopio de túnel de barrido (STM) y el microscopio de fuerza atómica (AFM).
Notas al pie
- 1 “Microscopio Electrónico de Transmisión JEM-ARM200F”, JEOL USA Inc, www.Jeolusa.com/products/tran... especificaciones. Accedido 28/08/2015.
Glosario
- microscopio de fuerza atómica
- un microscopio con sonda de barrido que utiliza una sonda delgada que se pasa justo por encima del espécimen para medir las fuerzas entre los átomos y la sonda
- binoculares
- que tiene dos oculares
- Microscopio de campo claro
- un microscopio óptico compuesto con dos lentes; produce una imagen oscura sobre un fondo brillante
- perilla de enfoque grueso
- una perilla en un microscopio que produce movimientos relativamente grandes para ajustar el enfoque
- cromóforos
- pigmentos que absorben y reflejan longitudes de onda particulares de la luz (dándoles un color)
- lente de condensador
- una lente en un microscopio que enfoca la luz de la fuente de luz sobre el espécimen
- microscopio confocal
- un microscopio láser de barrido que utiliza tintes fluorescentes y láseres de excitación para crear imágenes tridimensionales
- microscopio de campo oscuro
- un microscopio óptico compuesto que produce una imagen brillante sobre un fondo oscuro; típicamente un microscopio de campo claro modificado
- diafragma
- un componente de un microscopio; normalmente consiste en un disco debajo del escenario con orificios de varios tamaños; se puede ajustar para permitir que más o menos luz de la fuente de luz llegue al espécimen
- microscopio diferencial de interferencia-contraste
- un microscopio que utiliza luz polarizada para aumentar el contraste
- microscopio electrónico
- un tipo de microscopio que utiliza haces de electrones de longitud de onda corta en lugar de luz para aumentar el aumento y la resolución
- perilla de enfoque fino
- una perilla en un microscopio que produce movimientos relativamente pequeños para ajustar el enfoque
- microscopio de fluorescencia
- un microscopio que utiliza fluorocromos naturales o manchas fluorescentes para aumentar el contraste
- fluorocromos
- cromóforos que emiten fluorescencia (absorben y luego emiten luz)
- iluminador
- la fuente de luz en un microscopio
- inmunofluorescencia
- una técnica que utiliza un microscopio de fluorescencia y fluorocromos específicos de anticuerpos para determinar la presencia de patógenos específicos en un espécimen
- monocular
- tener un solo ocular
- lentes de objetivo
- en un microscopio óptico, las lentes más cercanas al espécimen, típicamente localizadas en los extremos de las torretas
- lente ocular
- en un microscopio, el cristalino más cercano al ojo (también llamado ocular)
- lente de inmersión de aceite
- una lente de objetivo especial en un microscopio diseñado para ser utilizado con aceite de inmersión para mejorar la resolución
- microscopio de contraste de fase
- un microscopio óptico que utiliza un tope anular y una placa anular para aumentar el contraste
- reóstato
- un regulador de intensidad que controla la intensidad del iluminador en un microscopio óptico
- Microscopio electrónico de barrido (SEM)
- un tipo de microscopio electrónico que rebota electrones del espécimen, formando una imagen de la superficie
- microscopio de la sonda de exploración
- un microscopio que utiliza una sonda que viaja a través de la superficie de un espécimen a una distancia constante mientras se mide la corriente, que es sensible al tamaño del espacio
- microscopio de túnel de exploración
- un microscopio que utiliza una sonda que se pasa justo por encima de la muestra ya que una polarización de voltaje constante crea el potencial de una corriente eléctrica entre la sonda y la muestra
- etapa
- la plataforma de un microscopio en la que se colocan los portaobjetos
- aumento total
- en un microscopio óptico es un valor calculado multiplicando el aumento del ocular por el aumento de las lentes del objetivo
- Microscopio electrónico de transmisión (TEM)
- un tipo de microscopio electrónico que utiliza un haz de electrones, enfocado con imanes, que pasa a través de un espécimen delgado
- Microscopio de dos fotones
- un microscopio que utiliza luz infrarroja o de longitud de onda larga para fluorescer fluorocromos en el espécimen
- Perillas mecánicas de escenario x-y
- perillas en un microscopio que se utilizan para ajustar la posición del espécimen en la superficie del escenario, generalmente para centrarlo directamente sobre la luz