7.3: Efectos estructurales en la acidez y basicidad
- Page ID
- 2362

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Now that we know how to quantify the strength of an acid or base, our next job is to gain an understanding of the fundamental reasons behind why one compound is more acidic or more basic than another. This is a big step: we are, for the first time, taking our knowledge or organic structure and applying it to a question of organic reactivity. Many of the ideas that we’ll see for the first here will continue to apply throughout the text as we tackle many other organic reaction types.
7.3A: Periodic trends
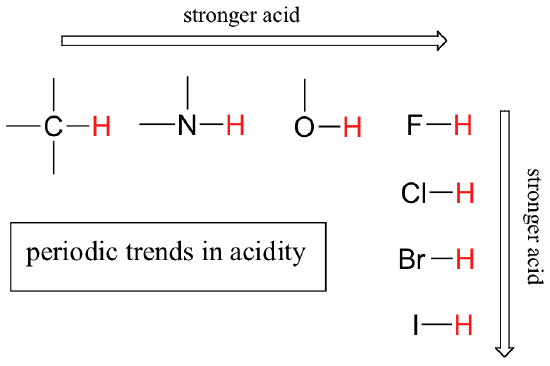
First, we will focus on individual atoms, and think about trends associated with the position of an element on the periodic table. We’ll use as our first models the simple organic compounds ethane, methylamine, and methanol, but the concepts apply equally to more complex biomolecules, such as the side chains of alanine, lysine, and serine.
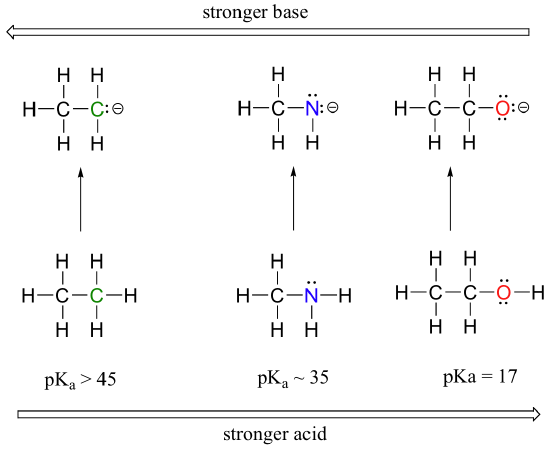
We can see a clear trend in acidity as we move from left to right along the second row of the periodic table from carbon to nitrogen to oxygen. The key to understanding this trend is to consider the hypothetical conjugate base in each case: the more stable (weaker) the conjugate base, the stronger the acid. Look at where the negative charge ends up in each conjugate base. In the ethyl anion, the negative charge is borne by carbon, while in the methylamine anion and methoxide anion the charges are located on a nitrogen and an oxygen, respectively. Remember the periodic trend in electronegativity (section 2.3A): it also increases as we move from left to right along a row, meaning that oxygen is the most electronegative of the three, and carbon the least. The more electronegative an atom, the better it is able to bear a negative charge. Thus, the methoxide anion is the most stable (lowest energy, least basic) of the three conjugate bases, and the ethyl anion is the least stable (highest energy, most basic).
We can use the same set of ideas to explain the difference in basicity between water and ammonia.
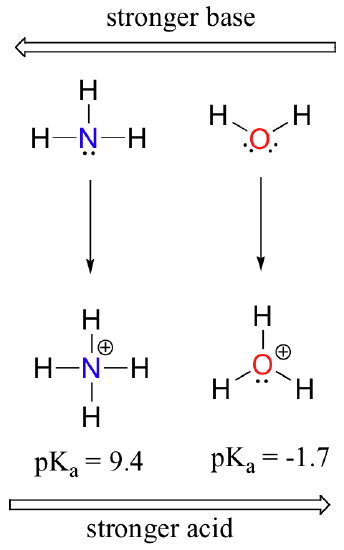
By looking at the pKavalues for the appropriate conjugate acids, we know that ammonia is more basic than water. Oxygen, as the more electronegative element, holds more tightly to its lone pair than the nitrogen. The nitrogen lone pair, therefore, is more likely to break away and form a new bond to a proton - it is, in other words, more basic. Once again, a more reactive (stronger) conjugate base means a less reactive (weaker) conjugate acid.
When moving vertically within a given column of the periodic table, we again observe a clear periodic trend in acidity. This is best illustrated with the halides: basicity, like electronegativity, increases as we move up the column.
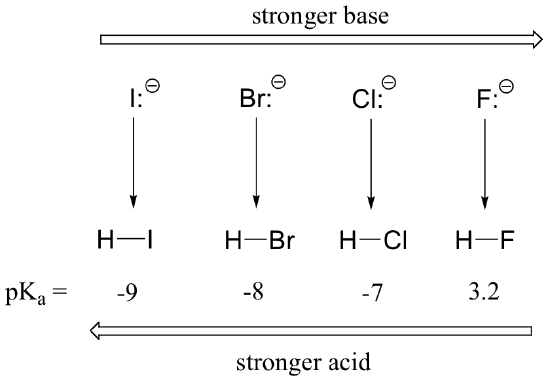
Conversely, acidity in the haloacids increases as we move down the column.
In order to make sense of this trend, we will once again consider the stability of the conjugate bases. Because fluorine is the most electronegative halogen element, we might expect fluoride to also be the least basic halogen ion. But in fact, it is the least stable, and the most basic! It turns out that when moving vertically in the periodic table, the size of the atom trumps its electronegativity with regard to basicity. The atomic radius of iodine is approximately twice that of fluorine, so in an iodine ion, the negative charge is spread out over a significantly larger volume:

This illustrates a fundamental concept in organic chemistry that is important enough to put in red:
Electrostatic charges, whether positive or negative, are more stable when they are ‘spread out’ than when they are confined to one atom.
We will see this idea expressed again and again throughout our study of organic reactivity, in many different contexts. For now, the concept is applied only to the influence of atomic radius on anion stability. Because fluoride is the least stable (most basic) of the halide conjugate bases, HF is the least acidic of the haloacids, only slightly stronger than acetic acid. HI, with a pKa of about -9, is one the strongest acids known.
More importantly to the study of biological organic chemistry, this trend tells us that thiols are more acidic than alcohols. The pKa of the thiol group on the cysteine side chain, for example, is approximately 8.3, while the pKa for the hydroxl on the serine side chain is on the order of 17.
To reiterate: acid strength increases as we move to the right along a row of the periodic table, and as we move down a column.
| Example 7.6 |
|---|
| Draw the structure of the conjugate base that would form if the compound below were to react with 1 molar equivalent of sodium hydroxide:
|
7.3B: The resonance effect
In the previous section we focused our attention on periodic trends - the differences in acidity and basicity between groups where the exchangeable proton was bound to different elements. Now, it is time to think about how the structure of different organic groups contributes to their relative acidity or basicity, even when we are talking about the same element acting as the proton donor/acceptor. The first model pair we will consider is ethanol and acetic acid, but the conclusions we reach will be equally valid for all alcohol and carboxylic acid groups.
Despite the fact that they are both oxygen acids, the pKa values of ethanol and acetic acid are very different. What makes a carboxylic acid so much more acidic than an alcohol? As before, we begin by considering the conjugate bases.
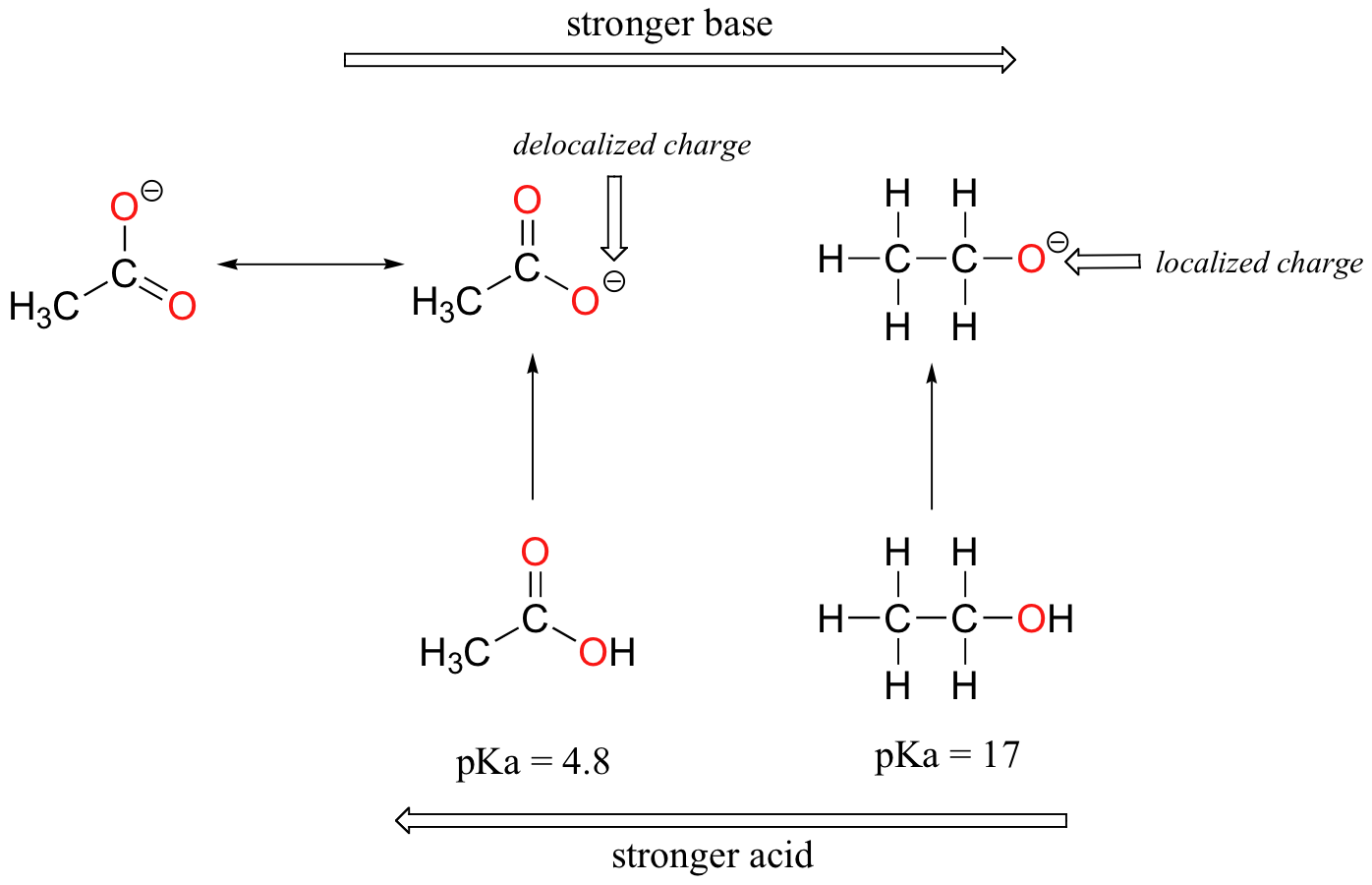
In both species, the negative charge on the conjugate base is held by an oxygen, so periodic trends cannot be invoked. For acetic acid, however, there is a key difference: a resonance contributor can be drawn in which the negative charge is localized on the second oxygen of the group. The two resonance forms for the conjugate base are equal in energy, according to our ‘rules of resonance’ (section 2.2C). What this means, you may recall, is that the negative charge on the acetate ion is not located on one oxygen or the other: rather it is shared between the two. Chemists use the term ‘delocalization of charge’ to describe this situation. In the ethoxide ion, by contrast, the negative charge is ‘locked’ on the single oxygen – it has nowhere else to go.
Now is the time to think back to that statement from the previous section that was so important that it got printed in bold font in its own paragraph – in fact, it is so important that we’ll just say it again: "Electrostatic charges, whether positive or negative, are more stable when they are ‘spread out’ than when they are confined to one atom." Now, we are seeing this concept in another context, where a charge is being ‘spread out’ (in other words, delocalized) by resonance, rather than simply by the size of the atom involved.
The delocalization of charge by resonance has a very powerful effect on the reactivity of organic molecules, enough to account for the difference of over 12 pKa units between ethanol and acetic acid (and remember, pKa is a log expression, so we are talking about a difference of over 1012 between the acidity constants for the two molecules). The acetate ion is that much more stable than the ethoxide ion, all due to the effects of resonance delocalization.
The resonance effect also nicely explains why a nitrogen atom is basic when it is in an amine, but not basic when it is part of an amide group. Recall that in an amide, there is significant double-bond character to the carbon-nitrogen bond, due to a second resonance contributor in which the nitrogen lone pair is part of a p bond.

While the electron lone pair of an amine nitrogen is ‘stuck’ in one place, the lone pair on an amide nitrogen is delocalized by resonance. Notice that in this case, we are extending our central statement to say that electron density – in the form of a lone pair – is stabilized by resonance delocalization, even though there is not a negative charge involved. Here’s another way to think about it: the lone pair on an amide nitrogen is not available for bonding with a proton – these two electrons are too ‘comfortable’ being part of the delocalized pi-bonding system. The lone pair on an amine nitrogen, by contrast, is not part of a delocalized p system, and is very ready to form a bond with any acidic proton that might be nearby.
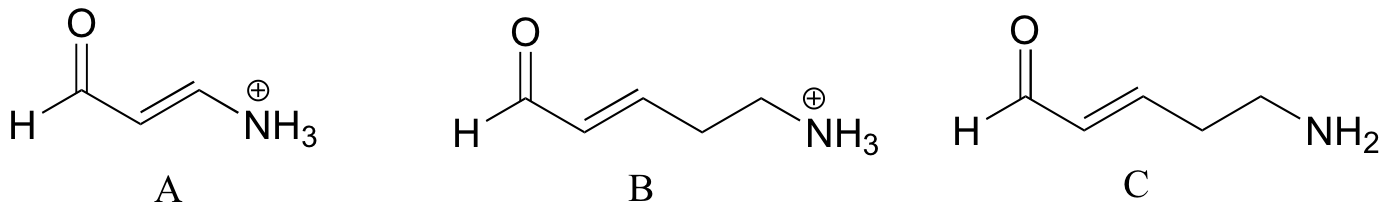
Often it requires some careful thought to predict the most acidic proton on a molecule. Ascorbic acid, also known as Vitamin C, has a pKa of 4.1.
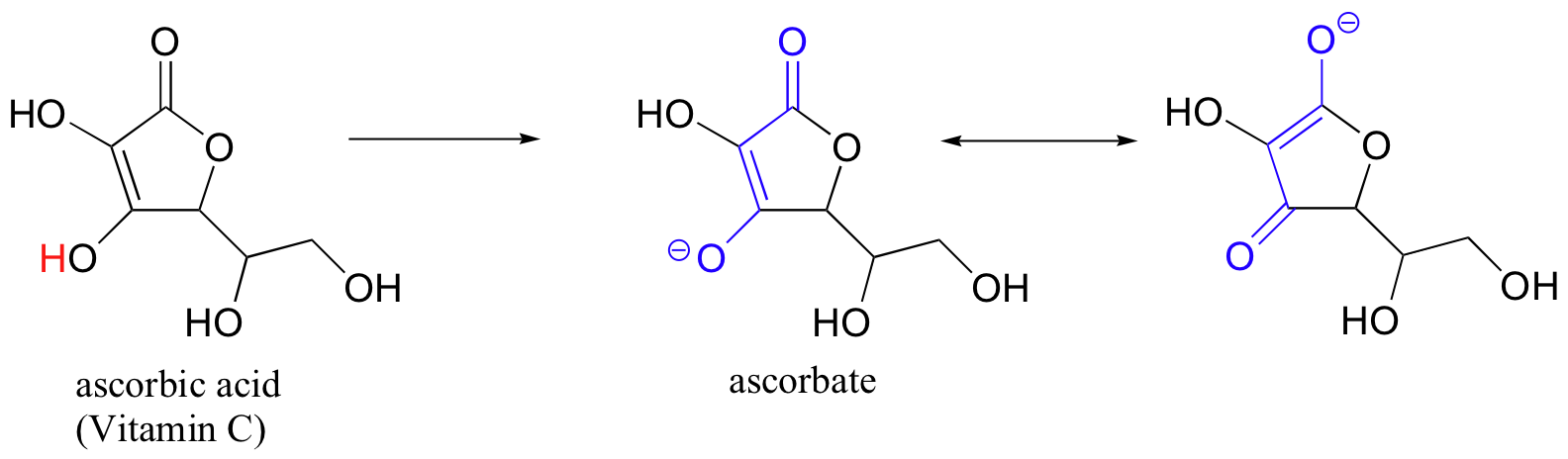
There are four hydroxyl groups on this molecule – which one is the most acidic? If we consider all four possible conjugate bases, we find that there is only one for which we can delocalized the negative charge over two oxygen atoms.
| Example 7.7 |
|---|
| Rank the compounds below from most acidic to least acidic, and explain your reasoning.
|
7.3C: The inductive effect
Compare the pKa values of acetic acid and its mono-, di-, and tri-chlorinated derivatives:
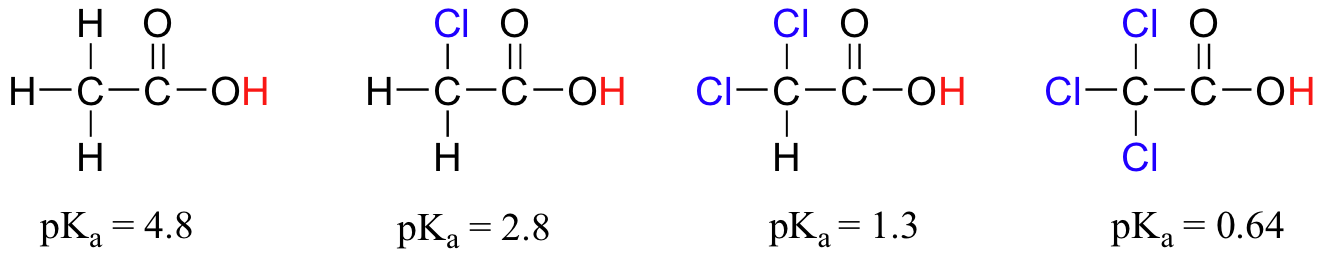
The presence of the chlorines clearly increases the acidity of the carboxylic acid group, but the argument here does not have to do with resonance delocalization, because no additional resonance contributors can be drawn for the chlorinated molecules. Rather, the explanation for this phenomenon involves something called the inductive effect. A chlorine atom is more electronegative than a hydrogen, and thus is able to ‘induce’, or ‘pull’ electron density towards itself, away from the carboxylate group. In effect, the chlorine atoms are helping to further spread out the electron density of the conjugate base, which as we know has a stabilizing effect. In this context, the chlorine substituent is called an electron-withdrawing group. Notice that the pKa-lowering effect of each chlorine atom, while significant, is not as dramatic as the delocalizing resonance effect illustrated by the difference in pKa values between an alcohol and a carboxylic acid. In general, resonance effects are more powerful than inductive effects.
The inductive electron-withdrawing effect of the chlorines takes place through covalent bonds, and its influence decreases markedly with distance – thus a chlorine two carbons away from a carboxylic acid group has a decreased effect compared to a chlorine just one carbon away.
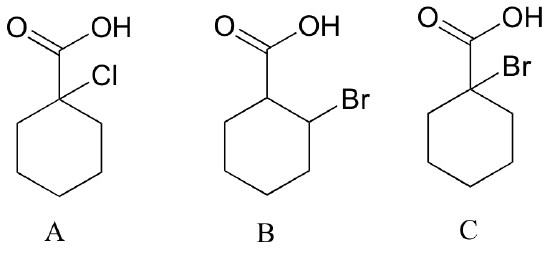
| Example 7.8 |
|---|
| Rank the compounds below from most acidic to least acidic, and explain your reasoning.
|