
1.9: Computación cristalográfica
- Page ID
- 77838

Los lectores que hayan llegado a este capítulo de manera secuencial notarán que, aparte del problema de fase, la relación entre el patrón de difracción (espacio recíproco) y la estructura cristalina (espacio directo) está mediada por una transformada de Fourier representado por la función de densidad electrónica:\(ρ(xyz)\) (ver el dibujo de la izquierda).
Los lectores también sabrán que la relación entre estos dos espacios es “holística”, es decir, que el valor de esta función, en cada punto de la celda unitaria de coordenadas\((xyz)\), es el resultado de “sumar” la contribución de “todos” factores de estructura [es decir, ondas difractadas en términos de sus amplitudes \(|F(hkl)|\)y fases\(Φ(hkl)\)] contenidas en el patrón de difracción. También recordarán que el patrón de difracción contiene muchos factores estructurales (varios miles para una estructura simple, y cientos de miles para una estructura proteica).
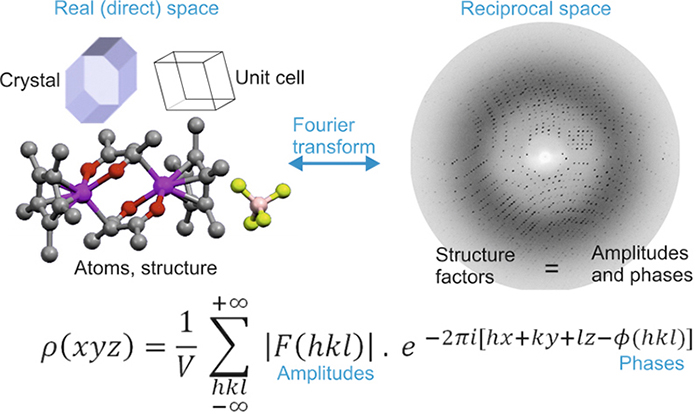
El “salto” entre espacios directos y recíprocos, mediado por una transformada de Fourier representada por la función de densidad electrónica
Además, el número de puntos en la celda unitaria, donde se tiene que calcular la función ρ, es muy alto. En una celda de aproximadamente 100 x 100 x 100 Angstrom 3, sería necesario calcular al menos 1000 puntos en cada dirección de celda unitaria para obtener una resolución de 100/1000, que equivale a 0.1 Angstrom en cada dirección. Esto significa calcular al menos 1000 x 1000 x 1000 = 1,000,000,000 puntos (mil millones de puntos) y en cada punto para “sumar” varios miles (o cientos de miles) factores de estructura F (hkl).
Por lo tanto, debe quedar claro que, independientemente de las dificultades del problema de fase, resolver una estructura cristalina implica el uso de computadoras.
Finalmente, el análisis de un cristal o estructura molecular implica también calcular muchos parámetros geométricos que definen distancias interatómicas, ángulos de enlace, ángulos de torsión, superficies moleculares, etc., utilizando las coordenadas atómicas (xyz).
El “hardware”
Por las razones descritas anteriormente, desde el inicio del uso de la Cristalografía como disciplina para determinar las estructuras moleculares y cristalinas, los cristalógrafos han dedicado especial atención al desarrollo de herramientas de cálculo para facilitar el trabajo cristalográfico. Con este objetivo, e incluso antes de que aparecieran las primeras computadoras, los cristógrafos introdujeron las llamadas “tiras Beevers-Lipson”, que fueron ampliamente utilizadas en todos los laboratorios de Cristalografía.


Las tiras Beevers-Lipson
Las tiras Beevers-Lipson (que eran tiras de papel que contenían los valores para algunas funciones trigonométricas) se utilizaron en laboratorios para acelerar los cálculos (a mano) de las transformaciones de Fourier (ver arriba: la función de densidad electrónica, por ejemplo).
Estas tiras fueron introducidas en 1936 por A.H. Beevers y H. Lipson. En la década de 1960, se distribuyeron más de 300 cajas a casi todos los laboratorios del mundo. También puedes echar un vistazo a la descripción realizada por la Unión Internacional de Cristalografía. La pesadilla fue mantener erguida esta caja, que tenía una base muy estrecha, de lo contrario era imposible mantener las tiras correctamente almacenadas!
Como era de esperar, la introducción de las primeras computadoras (o calculadoras electromecánicas) inspiró una gran esperanza en los cristalógrafos...


ENIAC (Electronic Numeric Integrator and Computer, 1945) — la primera computadora electrónica. Algunas fotos de las habitaciones donde se instaló.
ENIAC, abreviatura de Integrador Numérico Electrónico Y Computadora, fue la primera computadora electrónica de propósito general, cuyo diseño y construcción fueron financiados por el Ejército de Estados Unidos durante la Segunda Guerra Mundial. Fue la primera computadora digital capaz de ser reprogramada para resolver una amplia gama de problemas informáticos, especialmente el cálculo de mesas de tiro de artillería para el Laboratorio de Investigación Balística del Ejército de Estados Unidos.
El ENIAC tuvo importancia inmediata. Cuando se anunció en 1946, fue anunciado en la prensa como un “Cerebro Gigante”. Se jactó de velocidades mil veces más rápidas que las máquinas electromecánicas, un salto en la potencia informática que ninguna máquina ha igualado. Este poder matemático, aunado a la programabilidad de propósito general, entusiasmó a científicos e industriales.
Además de su velocidad, lo más destacable de ENIAC fue su tamaño y complejidad. ENIAC contaba con 17,468 tubos de vacío, 7,200 diodos de cristal, 1,500 relés, 70,000 resistencias, 10,000 capacitores y alrededor de 5 millones de juntas soldadas a mano. Pesaba 27 toneladas, era aproximadamente 2.6 m por 0.9 m por 26 m, ocupó 63 m² y consumió 150 kW de potencia.
Posteriormente, con el desarrollo de Electrónica y Microelectrónica, que introdujeron circuitos integrados, las computadoras se volvieron accesibles a los cristalógrafos, quienes acudieron en masa a estas instalaciones con grandes cajas de “tarjetas perforadas” (el único medio para el almacenamiento de datos en ese momento), conteniendo las intensidades de difracción y sus propios programas informáticos.

Una tarjeta perforada o una tarjeta perforada (o una tarjeta perforada o una tarjeta Hollerith o una tarjeta IBM), es un trozo de papel rígido que contiene información digital representada por la presencia o ausencia de agujeros en posiciones predefinidas. Fue utilizado por los cristalógrafos hasta finales de la década de 1970.


Cinta de papel perforada (mostrada en amarillo) y diferentes cintas magnéticas (así como algunos discos pequeños) utilizadas para el almacenamiento de datos durante las décadas de 1970 y 1980.
Alrededor de principios de la década de 1970, y durante más de una década, los cristalógrafos se convirtieron en una pesadilla para los gerentes y operadores de los llamados “centros de computación”, que funcionan en algunas universidades y centros de investigación.
En la década de 1980 los laboratorios de Cristalografía se “inundaron” de computadoras, lo que por primera vez dio a los cristalógrafos independencia de los grandes centros de computación. La serie de computadoras VAX (vendida por la compañía Digital Equipment Corporation) marcó una época espléndida para los cálculos cristalográficos. Permitieron el uso de cintas magnéticas y las primeras unidades de disco duro, con capacidad limitada (sólo unos cientos de MB) —muy grandes y pesadas, pero eliminaron la necesidad de las tediosas tarjetas perforadas. Nostálgicos deberían echar un vistazo a este enlace.!!!

Una computadora típica (de la serie VAX) utilizada en muchos laboratorios de Cristalografía durante la década de 1980.
A lo largo de los años, la computación cristalográfica se ha vuelto fácil y asequible gracias a las computadoras personales (PC), que satisfacen casi todas las necesidades de la mayoría de los cálculos cristalográficos convencionales, al menos concernientes a cristales de baja y media complejidad (hasta cientos de átomos). Su precio relativamente bajo y su capacidad para ensamblarse en “granjas” (para cálculo distribuido) proporcionan a los cristógrafos la mejor solución para casi cualquier tipo de cálculo.


Izquierda: Una computadora personal (PC) típica utilizada en la década de 2000
Derecha: Una granja de PC típica utilizada en la década de 2000
Sin embargo, la cristalografía aplicada a macromoléculas no solo necesita lo que podríamos llamar computación “dura”. El manejo de grandes mapas de densidad electrónica, que se utilizan para construir la estructura molecular de las proteínas, así como el posterior análisis estructural, requiere computadoras más sofisticadas con potentes procesadores gráficos y, si es posible, con la capacidad de mostrar imágenes tridimensionales utilizando gafas especializadas...

Una computadora Silicon Graphics utilizada para visualizar mapas y estructuras de densidad electrónica tridimensionales. El procesador y la pantalla se complementan con un transmisor de infrarrojos (caja negra en la pantalla) y las gafas utilizadas por el cristalógrafo.
Las instalaciones informáticas actuales representan un gran salto respecto a las capacidades disponibles a mediados del siglo XX, tal como se muestra en la representación del modelo estructural utilizado para la descripción estructural de la penicilina, basado en mapas tridimensionales de densidad electrónica... Y hasta mapas 3d donde también se usaron!...


Izquierda: Modelo tridimensional de la estructura de la penicilina, basado en el uso de mapas tridimensionales de densidad electrónica, como lo utilizó Dorothy C. Hodgkin , premio Nobel en 1964
Derecha: Representación de mapas de densidad electrónica 3D utilizados hasta mediados de la década de 1970. Los contornos son líneas de densidad electrónica y muestran las posiciones de los átomos individuales en la estructura
Una computadora personal típica comúnmente utilizada desde 2010 para cálculos cristalográficos y también por sus capacidades gráficas
El software
En la actualidad hay suficientes desarrollos de programas informáticos personales, institucionales o comerciales, o incluso instalaciones informáticas a través de servidores remotos, para satisfacer casi todas las necesidades de computación cristalográfica, así como muchas fuentes de las cuales se puede descargar la mayoría de esos programas. En este contexto, podría ser útil consultar los siguientes enlaces:
Programas de computadora cristalográficos
- Macromoléculas: El libro web del Departamento de Cristalografía y Biología Estructural (CSIC)
- De interés general: La lista de software cristalográfico mantenida por la Unión Internacional de Cristalografía - (IUCr)
Específicamente para compuestos de tamaño pequeño y mediano (moleculares o no) recomendamos usar el paquete Wingx que se puede descargar libremente por cortesía de Louis J. Farrugia, (Universidad de Glasgow, Reino Unido). Es fácil de instalar en una PC y contiene una interfaz que incluye los programas más importantes para problemas cristalográficos de tamaño pequeño y mediano. Además, para este tipo de compuestos existe un programa informático muy útil (Mercury), fácil de usar y gratuito, que incluye potentes gráficos y algunas otras herramientas analíticas para analizar estructuras cristalinas. Se puede descargar desde el Centro de Datos Cristalográficos de Cambridge, Reino Unido.
Los cristalógrafos de proteínas necesitan programas más específicos, y en este contexto recomendamos utilizar el enlace ofrecido por CCP4, Proyecto Computacional Colaborativo No. 4, Software para Cristalografía Macromolecular de Rayos X.
Por otro lado, el trabajo cristalográfico es actualmente inimaginable sin tener acceso a bases de datos cristalográficas, que contienen toda la información estructural que se está publicando y que tienen un claro valor agregado para el investigador. El tipo de estructura es lo que determina su inclusión en cualquiera de las bases de datos existentes. Así, los metales y compuestos intermetálicos se ponen a disposición en la base de datos CRYSTMET; los compuestos inorgánicos están centralizados en la base de datos ICSD (Inorganic Crystal Structure Database); orgánicos y organometálicos en CSD ( Cambridge Crystallographic Database); y proteínas en PDB (Protein Data Bank), que es un banco de datos (no una base de datos). Otras bases de datos, bancos de datos, etc., no necesariamente contienen información estructural en el sentido más preciso, pero también pueden ser muy útiles para los cristalógrafos. Y este es el caso de WebCite publicado por el Cambridge Crystalographic Data Centre (CCDC), que contiene más de 2000 artículos con información muy importante para la investigación de química estructural en su sentido más amplio, y en particular para el descubrimiento de fármacos farmacéuticos, el diseño de materiales o el desarrollo de fármacos, entre otros.
Bases de datos estructurales y bancos de datos
- CRISTMET: Metales y compuestos intermetálicos (licencia requerida)
- ICSD: Compuestos inorgánicos (licencia requerida)
- CSD: Compuestos orgánicos y organometálicos (licencia requerida)
- Glycosciences.de: Carbohidratos
- LipidBank: Lípidos
- PDB: Proteínas, Ácidos nucleicos y grandes complejos
- NDB: Ácidos nucleicos
Como se indicó, algunas de estas bases de datos (o bancos de datos) son públicas (Glycosciences.de, LipidBank, PDB y NDB), por lo que se pueden realizar búsquedas en línea. Sin embargo, otros (CRYSTMET, ICSD y CSD) requieren una licencia o incluso una instalación local.
Durante el periodo 1990-2012, CRYSTMET, ICSD y CSD han sido licenciados gratuitamente a todos los institutos de investigación del CSIC (CRYSTMET e ICSD) y a todas las instituciones académicas de España y países latinoamericanos (CSD). Sin embargo, debido a limitaciones económicas, las autoridades del CSIC decidieron reducir drásticamente este programa que se manejaba a través del Departamento de Cristalografía y Biología Estructural (en el Instituto de Química Física” Rocasolano”). Hoy en día este programa se mantiene de manera reducida, sólo para las instituciones españolas, como se puede apreciar a través de este enlace.