
1.8: El modelo estructural
- Page ID
- 77857

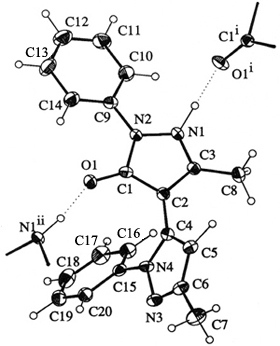
El análisis e interpretación de la función de densidad electrónica, es decir, la resolución de una estructura cristalina (molecular o no molecular) conduce a una distribución inicial de posiciones atómicas dentro de la celda unitaria que pueden ser representadas por puntos o pequeñas esferas:
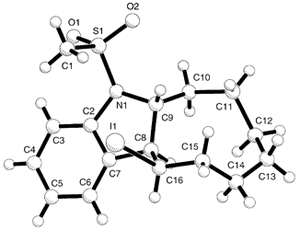

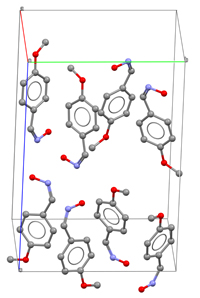
Una vez completado el modelo estructural, teniendo sentido estereoquímico e incluyendo su empaquetamiento cristalino, es necesario hacer uso de toda la información que podemos extraer de los datos experimentales, ya que el patrón de difracción generalmente contiene muchos más datos (intensidades) de los necesarios para localizar los átomos en sus coordenadas tridimensionales. Por ejemplo, para una estructura de tamaño mediano, con 50 átomos independientes en la unidad asimétrica (en la unidad estructural que se repite por las operaciones de simetría), el patrón de difracción generalmente contiene alrededor de 2500 factores de estructura, lo que implica aproximadamente 50 observaciones por átomo (cada átomo necesita 3 coordenadas). Sin embargo, para estructuras más complejas, como en el caso de las macromoléculas, la cantidad de datos experimentales disponibles normalmente no alcanza estos límites.
REFINAR EL MODELO FINAL
Los parámetros básicos asociados a una estructura tridimensional son, obviamente, las tres coordenadas posicionales (x, y, z) para cada átomo, dadas en términos de fracciones de células unitarias. Pero, en general, dada la sobredeterminación experimental mencionada anteriormente, el modelo atómico puede volverse más complejo. Por ejemplo, asociar cada átomo con un parámetro adicional que refleje su estado vibratorio térmico, en un primer acercamiento como una vibración térmica isotrópica (esférica) alrededor de su posición de equilibrio. Este nuevo parámetro se muestra normalmente en términos de diferente radio de la esfera que representa el átomo. Así, un modelo estructural isotrópico estaría representado por 4 variables por átomo: 3 posicionales + 1 térmica.
Sin embargo, para estructuras pequeñas y medianas (hasta varios cientos de átomos), el experimento de difracción suele contener datos suficientes para completar el modelo de vibración térmica, asociando un tensor (6 variables) a cada átomo que expresa el estado de vibración de manera anisotrópica, es decir, distinguiendo entre diferentes direcciones de vibración en forma de elipsoide (que se asemeja a la forma de una pelota de béisbol). Por lo tanto, un modelo anisotrópico cristalográfico requerirá 9 variables por átomo (3 posicionales + 6 vibracionales).
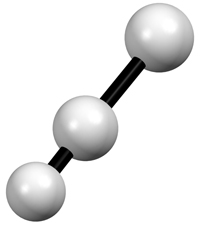
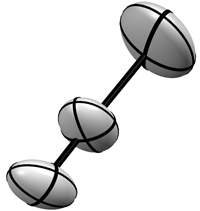
Izquierda: Tres átomos unidos representados con el modelo de vibración térmica isotrópica Derecha: Los mismos tres átomos mostrados a la izquierda, pero representados usando el modelo de vibración térmica anisotrópica

Izquierda: Modelo anisotrópico de la estructura tridimensional de una molécula, mostrando algunos átomos de moléculas vecinas.
Derecha: Modelo anisotrópico de la estructura tridimensional de una molécula que muestra su empaquetamiento cristalino.
Independientemente del tipo de modelo, isotrópico o anisotrópico, la sobreabundancia de datos experimentales antes mencionada permite una descripción del modelo estructural en términos de parámetros atómicos muy precisos (posicionales y vibracionales) que conducen a parámetros geométricos muy precisos de toda la estructura (interatómica distancias, ángulos de unión, etc.).
Este modelo refinado se obtiene mediante el método analítico de mínimos cuadrados. Mediante esta técnica, se permite que los átomos se “muevan” ligeramente de sus posiciones anteriores y se aplican factores térmicos a cada átomo de manera que el patrón de difracción calculado con este modelo es esencialmente el mismo que el experimental (observado), es decir, minimizando las diferencias entre el calculado y factores de estructura observados. Este proceso se lleva a cabo minimizando la función:
\[\sum w | |F_o| - |F_c| |^2 → 0\]
Función de mínimos cuadrados utilizada para refinar el modelo final de una estructura cristalina
donde\(w\) representa un factor de “peso” asignado a cada observación (intensidad), ponderando los efectos de las observaciones menos precisas frente a las más precisas y evitando posibles errores sistemáticos en las observaciones experimentales que podrían sesgar el modelo. Fo y Fc son factores de estructura observados y calculados, respectivamente.
Aunque por lo general la mencionada sobredeterminación experimental asegura el éxito de este proceso analítico de refinamiento, siempre debe ser controlado a través de los aspectos estereoquímicos, es decir, asegurando que los movimientos posicionales de los átomos sean razonables y que por lo tanto generen distancias dentro del valores esperados. De igual manera, los factores de vibración térmica (isotrópicos o anisotrópicos) asociados a los átomos deben mostrar siempre valores razonables.
Además del mencionado control de los cambios del modelo durante el proceso de refinamiento, parece obvio que (si todo va bien), adicionalmente el patrón de difracción calculado (F c) con el modelo refinado ( coordenadas + factores de vibración térmica) mostrarán una similitud creciente con el patrón observado (F o). La comparación entre ambos patrones (observados vs. calculados) se realiza a través del denominado\(R\) parámetro, que define el factor de “desacuerdo” entre los dos patrones:
\[R = \dfrac{\sum [ | |F_o| - |F_c| | ]}{|F_o|}\]
Factor de desacuerdo de un modelo estructural, calculado en términos de diferencias entre factores de estructura observados y calculados con el modelo final
El valor del factor de desacuerdo (R) se estima como un porcentaje (%), es decir, multiplicado por 100, de manera que las estructuras “bien” resueltas, con un grado apropiado de precisión, mostrarán un factor R por debajo de 0.10 (10%), lo que implica que el patrón calculado difiere de la observada (experimental) menos de 10%.
Los patrones de difracción de macromoléculas (enzimas, proteínas, etc.) generalmente no muestran una sobredeterminación tan grande de datos experimentales y por lo tanto es difícil alcanzar un modelo final anisotrópico. Además, en estos casos los valores del factor R son mayores que los de moléculas pequeñas y medianas, por lo que generalmente son aceptables valores alrededor o por debajo del 20%. Además, como resultado de esta relativa escasez de datos experimentales, el procedimiento analítico de refinamiento (mínimos cuadrados) debe combinarse con un proceso de modelado estereoquímico interactivo e imponiendo ciertas “restricciones suaves” a la geometría molecular.
VALIDACIÓN DE MODELOS
La confiabilidad de un modelo estructural tiene que ser evaluada en términos de varias pruebas, un procedimiento conocido como validación del modelo. Por lo tanto, el modelo estructural debe verificarse y validarse continuamente utilizando criterios estereoquímicos consistentes (por ejemplo, las longitudes de enlace y los ángulos de enlace deben ser aceptables). Por ejemplo, una distancia C-O de 0.8 Angstrom no sería aceptable para un grupo carbonilo (C = O). Del mismo modo, los ángulos de unión también deben ser consistentes con una geometría aceptable. Estos criterios son muy restrictivos para estructuras pequeñas o medianas, pero incluso en las estructuras de macromoléculas deben cumplir algunos criterios mínimos.

Valores máximos de dispersión generalmente aceptados para distancias interatómicas y ángulos de enlace en el modelo estructural de una macromolécula
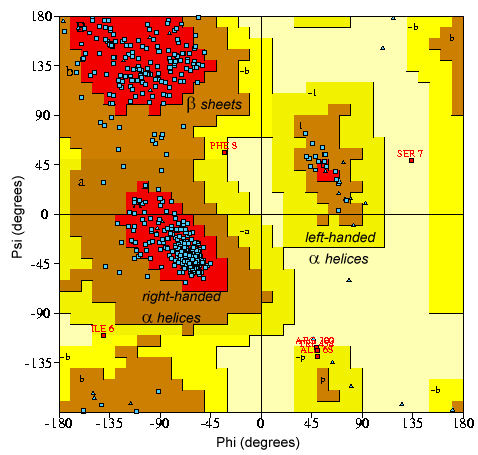
En el caso de las proteínas, el enlace peptídico (el enlace entre dos aminoácidos consecutivos) también debe satisfacer algunas restricciones geométricas. Los ángulos torsionales de este enlace no deben desviarse mucho de los valores aceptables de las conformaciones habituales que muestran las cadenas de aminoácidos, como se muestra en la denominada gráfica Ramachandran:
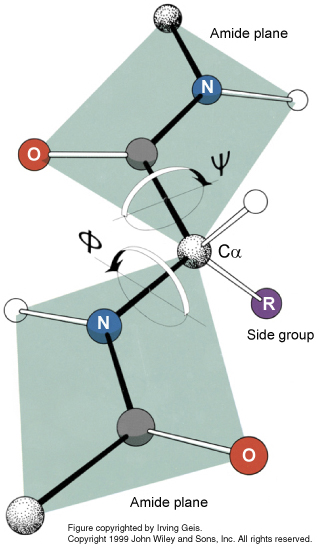
Izquierda: Representación esquemática del enlace peptídico, mostrando los dos ángulos de torsión (ψ y Φ) que lo definen.
Derecha: Gráfica Ramachandran que muestra las diferentes áreas permitidas (aceptables) para los ángulos torsionales de los enlaces peptídicos en una macromolécula. Las diferentes áreas dependen de los diferentes arreglos estructurales (hélices α-, β-láminas, etc.)
De igual manera, los valores de los factores térmicos asociados a cada átomo deben mostrar valores físicamente aceptables. Estos parámetros dan cuenta de la movilidad vibratoria térmica de las diferentes partes estructurales. Así, en la estructura de una macromolécula, estos valores deben ser consistentes con la ubicación interna o externa de la cadena, siendo generalmente menores para las partes internas, y mayores para las partes externas cercanas al disolvente.
GRADO DE CONFIABILIDAD DEL MODELO
Un modelo que ha sido “validado” de acuerdo con los criterios descritos anteriormente, es decir, que demuestra:
- un acuerdo razonable entre los factores de estructura observados y calculados,
- distancias de unión, ángulos de unión y ángulos de torsión que cumplen criterios estereoquímicos, y
- factores de vibración térmica físicamente razonables,
es un modelo confiable. Sin embargo, el concepto de confiabilidad no es un parámetro cuantitativo que pueda escribirse en términos de un solo número. Por lo tanto, para interpretar un modelo estructural hasta sus consecuencias lógicas hay que tener en cuenta que es solo una representación simplificada, extraída de una función de densidad electrónica:
\[\rho(x y z)=\frac{1}{V} \sum_{\substack{h k l}}^{+\infty}|F(h k l)| \cdot e^{-2 \pi i[h x+k y+l z-\phi(h k l)]}\]
sobre el que se han posicionado los átomos y que está siendo afectado por algunas condiciones descritas en otra sección, que le invitamos a leer.
Pero, en cualquier caso, el trabajo cristalográfico bien hecho siempre proporciona parámetros atómicos (posicionales y vibracionales) junto con sus estimaciones de precisión asociadas. Esto quiere decir que cualquier parámetro cristalográfico directo (coordenadas atómicas y factores de vibración) o derivado (distancias, ángulos, etc.) suele ser expresado por un número seguido de su desviación estándar (entre paréntesis) que afecta a la última figura. Por ejemplo, una distancia interatómica expresada como 1.541 (2) Angstroms significa una distancia de 1.541 y una desviación estándar de 0.002.
La configuración absoluta (o estereoquímica absoluta)
Como se dijo en un capítulo anterior, todas las moléculas o estructuras en las que no están presentes ni planos especulares ni centros de simetría, tienen una configuración absoluta, es decir, que son distintas de sus imágenes especulares (no pueden superponerse).
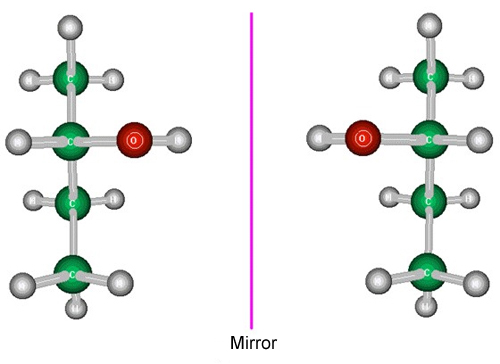
Modelos estructurales que muestran dos enantiómeros de un compuesto (las dos moléculas son imágenes especulares)
Estas diferencias estructurales particulares, muy importantes en lo que se refiere a las propiedades moleculares, se pueden determinar sin ambigüedades a través del experimento de difracción (sin utilizar ningún estándar externo). Esto se puede llevar a cabo utilizando el llamado efecto de dispersión anómalo que muestran los átomos cuando se utilizan longitudes de onda de rayos X apropiadas. Esta característica también se utiliza con mucho éxito como método para resolver el problema de fase de los cristales macromoleculares. No parece difícil entender que los enantiómeros moleculares tienen diferentes propiedades, ya que al final son moléculas diferentes, pero en cuanto a su actividad biológica (si la hay) la situación es particularmente llamativa.

Las moléculas enantioméricas que se representan en la figura izquierda fueron introducidas en el mercado por una compañía farmacéutica y, obviamente, mostraron diferentes propiedades.
Las propiedades de DARVON (Napsilato de Dextropropoxifeno) están disponibles a través de este enlace, mientras que se interrumpió la producción de NOVRAD (Napsilato de Levopropoxifeno).
La señal de difracción experimental que permite esta diferenciación estructural es consecuencia de que el factor de dispersión atómica no se comporta como un número real cuando la frecuencia de los rayos X es similar a la frecuencia natural de la absorción atómica. Véase también el capítulo dedicado a la dispersión anómala.
Bajo estas condiciones, ya no se cumple la Ley de Friedel y por lo tanto estructuran factores como |F h, k, l | y | F -h, -k, -l | será ligeramente diferente. Estas diferencias se evalúan en términos de los denominados estimadores Bijvoet, que comparan las proporciones para los factores de estructura observados para dichos pares de reflexión con las proporciones correspondientes para los factores de estructura calculados utilizando los dos posibles modelos absolutos. Sólo una de estas dos comparaciones mantendrá el mismo tipo de sesgo:
\[\frac{|F(h k l)|_{o}}{|F(\bar{h} \bar{k} \bar{l})|_{o}} \text { vs. } \frac{|F(h k l)|_{c}}{|F(\bar{h} \bar{k} \bar{l})|_{c}}\]

Comparación de ratios de Bijvoet - Johannes Martin Bijvoet (1892-1980)
Así, si el cociente entre los factores de estructura observados es <1, el mismo cociente para los factores de estructura calculados también debería ser <1. O, por el contrario, ambos cocientes deberían ser >1. Si esto es cierto para un gran número de pares de reflexión indicará que el modelo absoluto es el correcto. Si no es así, el modelo estructural tiene que ser invertido.
El lector interesado también debería echar un vistazo a las páginas web sobre dispersión anómala, preparadas por Ethan A. Merritt.
EL RESULTADO FINAL
La información que describe un modelo cristalográfico final está compuesta por:
- Datos del experimento de difracción: longitud de onda y patrón de difracción (la intensidad de miles o incluso cientos de miles de ondas difractadas con sus índices hkl),
- Dimensiones de celda unitaria derivadas del patrón de difracción (de la celda recíproca),
- La simetría presente en el cristal, derivada de la red recíproca (del patrón de difracción), y
- Posiciones atómicas (coordenadas y factores de vibración térmica) y, de ser necesario, el denominado factor poblacional, como se indica en la siguiente tabla.
Las posiciones atómicas suelen darse como coordenadas fraccionarias (fracciones de los ejes celulares unitarios), pero a veces, especialmente para macromoléculas donde la información suele referirse a la molécula aislada, se dan como coordenadas absolutas, es decir, expresadas en Angstrom y referidas a un sistema de ortogonales ejes independientes de los cristalográficos (ver abajo).

Información sobre varios átomos de una estructura proteica utilizando el llamado formato PDB (Protein Data Bank), es decir, coordenadas atómicas en Angstrom sobre un sistema de ejes ortogonales, diferentes a los cristalográficos. Para mayor claridad, se han omitido las desviaciones estándar estimadas.
El factor poblacional es la fracción de átomo ubicada en una posición específica, aunque este factor suele ser 1. El significado de este parámetro requiere una explicación para el principiante, ya que podría entenderse que los átomos podrían dividirse en partes, lo que obviamente no tiene significado físico. Debido a las vibraciones atómicas, y a que el experimento de difracción tiene una duración en el tiempo, es posible que en algunas de las celdas unitarias falten átomos. Así, en lugar de una ocupación completa (factor poblacional = 1), el sitio correspondiente, en una celda unitaria promedio, contendrá sólo una fracción del átomo. En estos casos se dice que la red cristalina tiene defectos y los factores de población menores a 1 reflejan una fracción de celdas unitarias donde se ocupa una posición atómica específica. Obviamente, una fracción de células unitarias donde la misma posición está vacía complementa el factor poblacional a la unidad. Por lo tanto, el modelo cristalográfico refleja la estructura promedio de todas las células unitarias durante el tiempo del experimento.
Las coordenadas atómicas y en general toda la información recopilada de un estudio cristalográfico, se almacenan en bases de datos accesibles. Existen distintas bases de datos, dependiendo del tipo de compuesto o molécula, pero esto se discutirá en otro capítulo de estas páginas.
REPRESENTACIONES GRÁFICAS DEL MODELO
El modelo estructural final (coordenadas atómicas, factores térmicos y, posiblemente, factores de población) proporciona directamente información adicional que conduce a un conocimiento detallado de la propia estructura, incluyendo longitudes de enlace, ángulos de enlace, ángulos de torsión, planos moleculares, impulso dipolar, etc., y cualquier otro detalle estructural que podría ser útil para comprender la funcionalidad y/o propiedades del material en estudio.

En el caso de moléculas biológicas complejas, el uso de procesadores gráficos de alta calidad y modelos relativamente simples, facilita enormemente la comprensión de la relación entre estructura y función, como se muestra en la figura de la izquierda.
En la actualidad las técnicas computacionales y gráficas disponibles nos permiten obtener modelos hermosos y muy descriptivos que ayudan a visualizar y comprender estructuras, como se muestra en los siguientes ejemplos:
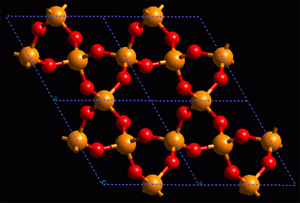
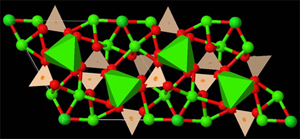
Izquierda: Modelo de bolas y palos para representar la estructura de un simple compuesto inorgánico. Derecha: Representación de un compuesto inorgánico, en el que se ha agregado una representación poliédrica parcial
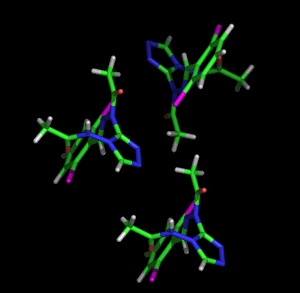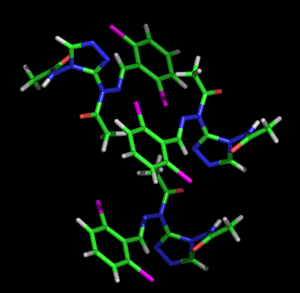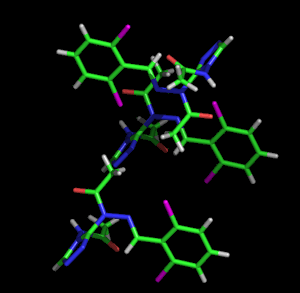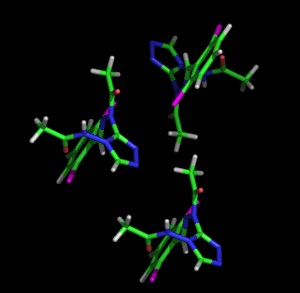
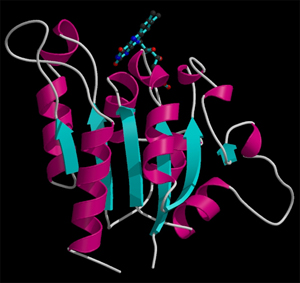
Izquierda: Modelo animado de sitcks para representar el empaque y la estructura molecular de un compuesto orgánico simple. Derecha: Dada la complejidad de las moléculas biológicas, los modelos que las representan suelen ser simples, mostrando el plegamiento general y los diferentes motivos estructurales (hélices α-hélices, β-hebras, bucles, etc.) mostrados con el modelo de cinta. El ejemplo también muestra una representación en barra de un cofactor ligado a la enzima.
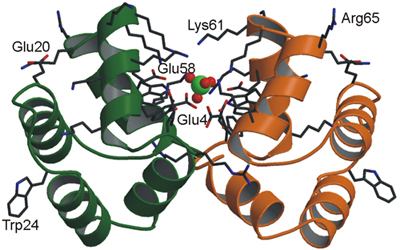
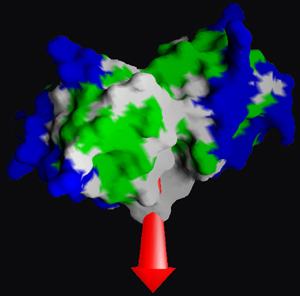
Izquierda: Modelo combinado de cintas y barras para representar la estructura dímera de una proteína que también muestra un ion sulfato en el medio, representado con bolas Derecha: Representación de la superficie de una molécula biológica donde los colores representan diferentes propiedades de la hidrofobia. La flecha representa el momento dipolar de la molécula.
Finalmente, utilizando información adicional de otras técnicas (como la crio-microscopía electrónica), o combinando dos conformaciones cristalinas diferentes de una molécula, se encuentran disponibles otros modelos como se muestra a continuación. Además, utilizando los tiempos de exposición ultracortos de los rayos X producidos por los láseres de electrones libres (European XFEL), los cristalógrafos son capaces de recolectar datos de difracción de macromoléculas en diferentes conformaciones, es decir, durante el transcurso de la realización de sus respectivas tareas. De esta manera, utilizando una gran cantidad de instantáneas de rayos X podemos producir como una película donde podemos seguir las modificaciones moleculares y por lo tanto entender su función.
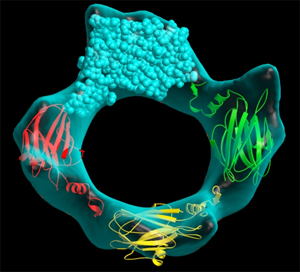

Izquierda: Modelo combinado de la estructura molecular de una proteína y una envoltura (obtenida por microscopía electrónica de alta resolución) que muestra un poro formado por la asociación de cuatro moléculas de proteína Derecha: Modelo animado simplificado que muestra el plegamiento de la columna vertebral de un enzima y los cambios estructurales entre dos estados moleculares: activo (abierto) e inactivo (cerrado). Las estructuras de ambos estados fueron determinadas por cristalografía