
30.10: La superficie de energía potencial se puede calcular usando mecánica cuántica
- Page ID
- 80205
Una superficie de energía potencial (PES) describe la energía potencial de un sistema, especialmente una colección de átomos, en términos de ciertos parámetros, normalmente las posiciones de los átomos. La superficie podría definir la energía en función de una o más coordenadas; si solo hay una coordenada, la superficie se denomina curva de energía potencial o perfil de energía. Es útil utilizar la analogía de un paisaje: para un sistema con dos grados de libertad (por ejemplo, dos longitudes de enlace), el valor de la energía (analogía: la altura de la tierra) es función de dos longitudes de enlace (analogía: las coordenadas de la posición en el suelo). La Superficie de Energía Potencial representa el concepto de que cada geometría (tanto externa como interna) de los átomos de las moléculas en una reacción química le ha asociado una energía potencial única. Esto crea un “paisaje” energético suave y la química se puede ver desde una perspectiva topológica de partículas que evolucionan a medida que pasan a través de “valles” y “pases” de energía potencial.
El concepto PES encuentra aplicación en campos como la química y la física, especialmente en las subramas teóricas de estas materias. Se puede utilizar para explorar teóricamente las propiedades de estructuras compuestas por átomos, por ejemplo, encontrar la forma de energía mínima de una molécula o calcular las velocidades de una reacción química.
Curvas de energía potencial (superficies de energía potencial 2-D)
La energía de un sistema de dos átomos depende de la distancia entre ellos. A grandes distancias la energía es cero, lo que significa “sin interacción”. A distancias de varios diámetros atómicos dominan las fuerzas atractivas, mientras que a aproximaciones muy cercanas la fuerza es repulsiva, haciendo que la energía suba. Los efectos atractivos y repulsivos se equilibran en el punto mínimo de la curva. Las gráficas que ilustran esta relación son bastante útiles para definir ciertas propiedades de un enlace químico (Figura\(\PageIndex{1}\)).

La distancia internuclear a la que se produce el mínimo de energía potencial define la longitud del enlace. Esto se conoce más correctamente como la longitud del enlace de equilibrio porque el movimiento térmico hace que los dos átomos vibren alrededor de esta distancia. En general, cuanto más fuerte es el enlace, menor es la longitud del enlace.
Las fuerzas atractivas operan entre todos los átomos, pero a menos que el mínimo de energía potencial sea al menos del orden de RT, los dos átomos no podrán resistir la influencia disruptiva de la energía térmica el tiempo suficiente para dar como resultado una molécula identificable. Así podemos decir que existe un enlace químico entre los dos átomos en H 2. La débil atracción entre los átomos de argón no permite que Ar 2 exista como molécula, pero sí da lugar a la fuerza v an der Waals que mantiene unidos los átomos de argón en sus formas líquida y sólida.
Energía potencial y teoría cuántica de energía cinética nos dicen que un electrón en un átomo posee energía cinética así\(K\) como energía potencial\(V\), por lo que la energía total\(E\) es siempre la suma de los dos:\(E = V + K\). La relación entre ellos es sorprendentemente simple:\(K = –0.5 V\). Esto significa que cuando se forma un enlace químico (un proceso exotérmico con\(ΔE < 0\)), la disminución de la energía potencial va acompañada de un aumento en la energía cinética (encarnada en el impulso de los electrones de unión), pero la magnitud de este último cambio es solo la mitad, por lo que el cambio en el potencial la energía siempre domina. La energía de enlace\(–ΔE\) tiene la mitad de la magnitud de la disminución de la energía potencial.
Definición Matemática y Computación de una Superficie Energética Potencial
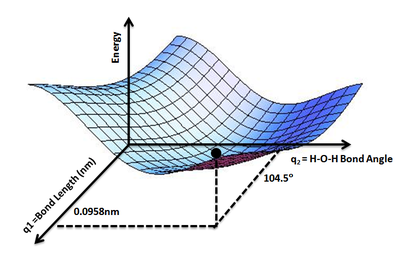
La geometría de un conjunto de átomos puede ser descrita por un vector, r, cuyos elementos representan las posiciones de los átomos. El vector\(r\) podría ser el conjunto de las coordenadas cartesianas de los átomos, o también podría ser un conjunto de distancias y ángulos interatómicos. Dado\(r\), la energía en función de las posiciones,\(V(r)\), es el valor de\(V(r)\) para todos los valores\(r\) de interés. Usando la analogía del paisaje de la introducción,\(V(r)\) da la altura sobre el “paisaje energético” para que surja el concepto de una superficie energética potencial. Un ejemplo es el PES para molécula de agua (Figura\(\PageIndex{2}\)) que muestra el mínimo de energía correspondiente a la estructura molecular optimizada para longitudes de enlace agua-O-H de 0.0958 nm y un ángulo de enlace H-O-H de 104.5°
Para definir la ubicación de un átomo en el espacio tridimensional se requieren tres coordenadas (por ejemplo,\(x\),\(y\), y\(z\) o\(r\),\(\theta\) y\(phi\) en coordenadas cartesianas y esféricas) o grados de libertad. Sin embargo, una reacción y por lo tanto los PES correspondientes no dependen de la posición absoluta de la reacción, solo de las posiciones relativas (grados internos). De ahí que se pueda eliminar tanto la traslación como la rotación de todo el sistema (cada uno con 3 grados de libertad, asumiendo geometrías no lineales). Entonces la dimensionalidad de un PES es
\[3N-6\nonumber \]
donde\(N\) es el número de átomos que implica en la reacción, es decir, el número de átomos en cada reactivo). El PES es una hipersuperficie con muchos grados de libertad y típicamente solo unos pocos se trazan en cualquier momento para su comprensión. Consulte Calcular el número de modos vibracionales para obtener una imagen más detallada de cómo se aplica esto al cálculo del número de vibraciones en una molécula
Para estudiar una reacción química utilizando el PES en función de las posiciones atómicas, es necesario calcular la energía para cada disposición atómica de interés. Los métodos para calcular la energía de una disposición atómica particular de átomos son bien conocidos. Para sistemas químicos muy simples o cuando se hacen aproximaciones simplificadas sobre interacciones interatómicas, a veces es posible utilizar una expresión derivada analíticamente para la energía en función de las posiciones atómicas. Un ejemplo es
\[\ce{H + H_2 -> H_2 + H}\label{30.10.1} \]
un sistema que se describe por una función de las tres\(\ce{H-H}\) distancias. Para sistemas más complicados, el cálculo de la energía de una disposición particular de átomos suele ser demasiado costoso desde el punto de vista computacional para que las representaciones a gran escala de la superficie sean factibles.
Aplicaciones de Superficies Energéticas Potenciales
Un PES es una herramienta conceptual para ayudar en el análisis de geometría molecular y dinámica de reacción química. Una vez evaluados los puntos necesarios en un PES, los puntos pueden clasificarse de acuerdo con la primera y segunda derivadas de la energía con respecto a la posición, que son respectivamente el gradiente y la curvatura. Los puntos estacionarios (o puntos con gradiente cero) tienen un significado físico: los mínimos de energía corresponden a especies químicas físicamente estables y los puntos de sillín corresponden a estados de transición, el punto de energía más alto en la coordenada de reacción (que es la vía de energía más baja que conecta una sustancia química reactivo a un producto químico).
Una reacción endotérmica hipotética PES
La figura\(\PageIndex{3}\) muestra un ejemplo de un PES para un sistema de reacción hipotético, con el correspondiente mapa de contorno de energía 2-D proyectado en el plano inferior. El mapa de contorno muestra isolíneas de energía potencial, utilizando el color para diferenciar entre energías altas y bajas. En esta figura, la mayor energía potencial se indica con rojo, y a medida que el color cambia a lo largo de la escala ROYGBIV, la energía potencial disminuye hasta alcanzar el violeta, indicando la energía potencial más baja. En el PES, el\(z\) eje representa la energía potencial, con la energía del plano en el PES definida como 0 kJ. El esquema de color del PES tiene el mismo significado que el del mapa de contorno 2-D: el rojo es energía de alto potencial y el violeta es baja energía potencial.

Antes de la reacción, los reactivos se encuentran en (o cerca) del mínimo de energía a la derecha. (El pozo profundo en el PES y el óvalo más pequeño, violeta en el mapa de contorno.) A medida que avanza la reacción, los reactivos se muestran siguiendo la ruta de energía mínima hacia los productos, como se designa por la línea roja discontinua. El punto “2" designa el estado de transición en el punto de sillín, el punto de energía más alto del proceso de reacción. La reacción luego pasa a formar los productos, que se asientan en el pozo de energía potencial a la izquierda. (Designado por el pozo menos profundo en el PES y por el óvalo azul oscuro a la izquierda en el mapa de contorno.) Debido a que la energía potencial de los reactivos es mayor que la energía potencial de los productos, esta reacción es una reacción endotérmica. El punto “1" representa un segundo punto de sillín de mayor energía que es el estado de transición para una reacción alternativa que es posible para este conjunto de químicos.
La reacción de intercambio H A + H B H C ⇒ H A H B + H C
Una aplicación específica de un PES es el mapeo de la reacción mostrada en la ecuación\(\ref{30.10.1}\), el intercambio de átomos de hidrógeno en una molécula H 2. En este mapa, los átomos de H individuales están etiquetados como
\[H_A + H_BH_C \rightarrow H_AH_B + H_C \nonumber \]
Debemos tomar en cuenta el ángulo de colisión entre el\(H_A\) átomo y la\(H_BH_C\) molécula. Si fijamos este ángulo de colisión a 180°, podemos trazar un PES que depende de los dos parámetros de\(R_{BC}\) y\(R_{AB}\). El PES (figura\(\PageIndex{4a}\)) resultante y el mapa de contorno energético (figura\(\PageIndex{4b}\)) nos muestran que si las partículas están lo suficientemente separadas, la energía potencial del sistema de reacción comienza siendo descrita por la curva de energía potencial para la\(H_BH_C\) molécula, y termina siendo descrita por el potencial curva de energía de la\(H_AH_B \) molécula. Individualmente, ambas curvas tienen apariencias similares a la curva mostrada en la Figura\(\PageIndex{1}\). El PES y el mapa de contorno energético muestran la naturaleza simétrica de los cambios de energía potenciales que ocurren en este proceso de intercambio que involucra productos que son equivalentes a los reactivos.


A medida que el\(H_A\) átomo se acerca más de cerca a la\(H_BH_C\) molécula, las interacciones entre estas partículas comienzan a afectar la energía potencial del sistema. Hay muchas vías posibles de energía potencial que la reacción podría seguir. La figura\(\PageIndex{5}\) muestra el mapa de contorno de energía para tres posibles vías de reacción:

En la trayectoria 1, la longitud de\(H_BH_C\) unión\(R_{BC} \) se mantiene constante a medida que\(R_{AB}\) disminuye la distancia. Este tipo de interacción conduciría a una energía potencial cada vez mayor para el sistema a medida que el\(H_A\) átomo se acerca cada vez más a la\(H_BH_C\) molécula. Eventualmente, la nueva\(H_AH_B\) molécula se formaría, y el\(H_C\) átomo se rompería y se movería cada vez más lejos.
La ruta 2 muestra una segunda vía de reacción posible en la que la longitud del\(H_BH_C\) enlace\(R_{BC} \) aumenta a pesar de que el\(H_A\) átomo todavía está relativamente lejos. Esta vía es poco probable porque requiere una gran cantidad de energía potencial para estirar el\(H_BH_C\) enlace antes de que la fuerza de atracción del\(H_A\) átomo influya en este alargamiento del enlace.
A medida que las partículas viajan a lo largo de la Vía 3, los reactivos aún deben pasar a través de un máximo de energía potencial en el punto de sillín, pero este máximo local es la barrera de energía más baja que separa los reactivos de los productos. Como se señaló anteriormente, este punto de sillín se denomina estructura de estado de transición (designada por el punto rojo en la ruta 3). Esta es la estructura que está igualmente preparada para regresar a los reactivos o avanzar para formar productos. La vía 3 es la vía de energía mínima y, por lo tanto, la que tiene más probabilidades de seguirse durante una reacción exitosa.
A veces es útil crear mapas de contorno de energía que incluyan información sobre el estado vibratorio de los reactivos y productos, como se muestra en la figura\(\PageIndex{6}\).

La Reacción de Intercambio F + D 2 ⇒ DF + D
Al modelar la energía potencial para la reacción
\[\ce{F(g) + D_2(g) -> DF(g) + D(g)}\nonumber \]
es útil diferenciar entre los dos átomos de deuterio, para que podamos designarlos como\(\ce{D}_A\) y\(\ce{D}_B\). Hacerlo nos permite determinar cómo la energía potencial del sistema de pre-reacción se ve afectada por la\(\ce{D}_A\) distancia\(\ce{F}\) a\(R_{\ce{D}_A \ce{F}}\) y la\(\ce{D}_A\)\(\ce{D}_B\) distancia\(R_{\ce{D}_A \ce{D}_B} \) (figura\(\PageIndex{7}\)).

A medida que el\(\ce{F}\) átomo se acerca más de cerca a la\(\ce{D_2}\) molécula, las interacciones entre estas partículas comienzan a afectar la energía potencial del sistema. Hay muchas vías posibles de energía potencial que la reacción podría seguir. La figura\(\PageIndex{8}\) muestra el mapa de contorno de energía para tres posibles vías de reacción:

En la trayectoria 1, la longitud de\(\ce{D_2}\) unión\(R_{\ce{D}_A \ce{D}_B} \) se mantiene constante a medida que\(R_{\ce{D}_A \ce{F}}\) disminuye la distancia. Este tipo de interacción conduciría a una energía potencial cada vez mayor para el sistema a medida que el\(\ce{F}\) átomo se acerca cada vez más a la\(\ce{D_2}\) molécula. Eventualmente, la nueva\(\ce{D}_A \ce{F}\) molécula se formaría, y el\(\ce{D}_B\) átomo se rompería y se movería cada vez más lejos.
La ruta 2 muestra una segunda vía de reacción posible en la que la longitud del\(\ce{D_2}\) enlace\(R_{\ce{D}_A \ce{D}_B} \) aumenta a pesar de que el átomo F todavía está relativamente lejos. Esta vía es poco probable porque requiere una gran cantidad de energía potencial para estirar el\(\ce{D_2}\) enlace antes de que la fuerza de atracción del átomo F influya en este alargamiento del enlace.
A medida que las partículas viajan a lo largo de la Vía 3, los reactivos aún deben pasar a través de un máximo de energía potencial en el punto de sillín, pero este máximo local es la barrera de energía más baja que separa los reactivos de los productos. Como se señaló anteriormente, este punto de sillín se denomina estructura de estado de transición (designada por el punto rojo en la ruta 3, etiquetada como\(3^{\ddagger}\)). Esta es la estructura que está igualmente preparada para regresar a los reactivos o avanzar para formar productos. La vía 3 es la vía de energía mínima y, por lo tanto, la que tiene más probabilidades de seguirse durante una reacción exitosa.
Si tuviéramos que trazar la curva de energía potencial para la vía de reacción a lo largo de la ruta de energía mínima, obtendríamos la curva de energía potencial familiar de una reacción similar a la que se muestra en la Figura\(\PageIndex{9}\).

Colaboradores y Atribuciones
- Wikipedia
Stephen Lower, Professor Emeritus (Simon Fraser U.) Chem1 Virtual Textbook
- Tom Neils, Grand Rapids Community College