
5.1: ¿De qué trata la química teórica?
- Page ID
- 70814

La ciencia de la química trata de moléculas incluyendo los radicales, cationes y aniones que producen cuando se fragmentan o ionizan. Los químicos estudian moléculas aisladas (por ejemplo, como ocurren en la atmósfera y en ambientes astronómicos), soluciones de moléculas o iones disueltos en solventes, así como materiales sólidos, líquidos y plásticos compuestos por moléculas. Todas esas formas de materia molecular son de lo que se trata la química. La ciencia química incluye cómo hacer moléculas (síntesis), cómo detectarlas y cuantificarlas (análisis), cómo sondear sus propiedades y los cambios que sufren a medida que ocurren reacciones (físicas).
Estructura molecular: unión, formas, estructuras electrónicas
Uno de los temas más fundamentales que aborda la química es la estructura molecular, lo que significa cómo los átomos de la molécula están unidos entre sí por enlaces y cuáles son las distancias y ángulos interatómicos. Otro componente del análisis de estructura se relaciona con lo que están haciendo los electrones en la molécula; es decir, cómo se ocupan los orbitales de la molécula y en qué estado electrónico existe la molécula. Por ejemplo, en la molécula de arginina mostrada en la Figura 5.1, un grupo ácido\(HOOC^-\) carboxílico (sus átomos de oxígeno se muestran en rojo) está unido a un átomo de carbono adyacente (amarillo) que a su vez está unido a un grupo\(–NH_2\) amino (cuyo átomo de nitrógeno es azul). También conectados al átomo de carbono a están una cadena de tres\(–CH_2^-\) grupos metileno, un\(–NH^-\) grupo, luego un átomo de carbono unido tanto por un doble enlace a un\(–NH\) grupo imina como a un\(–NH_2\) grupo amino.
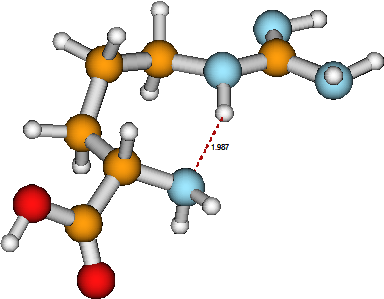
La conectividad entre los átomos en arginina viene dictada por las conocidas preferencias de valencia mostradas por los átomos de H, C, O y N. Los ángulos de enlace internos están, en gran medida, también determinados por las valencias de los átomos constituyentes (es decir, la\(sp_3\) o\(sp_2\) naturaleza de los orbitales de enlace). Sin embargo, existen otras interacciones entre los diversos grupos funcionales en la arginina que también contribuyen a su estructura final. En particular, el enlace de hidrógeno que une el átomo de nitrógeno del grupo a-amino al átomo de hidrógeno del\(–NH^-\) grupo hace que esta molécula se pliegue en una estructura menos extendida de lo que podría ser de otra manera.
¿Qué tiene que ver la teoría con cuestiones de estructura molecular y por qué es tan importante el conocimiento de la estructura? Es importante porque la estructura de una molécula tiene un papel muy importante en la determinación de los tipos de reacciones que sufrirá esa molécula, qué tipo de radiación absorberá y emitirá, y a qué sitios activos en moléculas vecinas o materiales cercanos se unirá. La forma de una molécula (por ejemplo, en forma de varilla, plana, globular, etc.) es una de las primeras cosas en las que piensa un químico cuando intenta predecir dónde, en otra molécula o en una superficie o en una membrana celular, la molécula encajará y podrá unirse y tal vez reaccionar. La presencia de pares solitarios de electrones (que actúan como sitios base de Lewis), de\(\pi\) orbitales (que pueden actuar como sitios donadores de electrones y aceptores de electrones), y de grupos altamente polares o iónicos guían al químico aún más para determinar en qué parte del marco de la molécula varias especies reaccionantes (por ejemplo, electrófilo o nucleófilo o radical) serán los más fuertemente atraídos. Claramente, la estructura molecular es un aspecto crucial de la caja de herramientas de los químicos.
¿Cómo se relaciona la teoría con la estructura molecular? Como discutimos en la Parte 1 de este texto, la aproximación Born-Oppenheimer nos lleva a utilizar la mecánica cuántica para predecir la energía\(E\) de una molécula para cualquier posición ({\(R_a\)}) de sus núcleos, dado el número de electrones Ne en la molécula (o ion). Esto significa, por ejemplo, que la energía de la molécula de arginina en su estado electrónico más bajo (es decir, con los electrones ocupando los orbitales de menor energía) puede determinarse para cualquier ubicación de los núcleos si se puede resolver la ecuación de Schrödinger que gobierna los movimientos de los electrones.
Si no has tenido una buena clase sobre cómo se usa la mecánica cuántica dentro de la química, te insto a que te tomes el tiempo necesario para dominar la Parte 1. En esas páginas, presento los conceptos centrales de la mecánica cuántica y muestro cómo se aplican a varios casos muy importantes incluyendo
- electrones que se mueven en dimensiones 1, 2 y 3 y cómo estos modelos se relacionan con estructuras electrónicas de polienos y con bandas electrónicas en sólidos
- las densidades de probabilidad clásica y cuántica y en qué se diferencian,
- propagación de tiempo de las funciones de onda cuántica,
- el Hückel o modelo de unión química de unión estrecha entre orbitales atómicos,
- vibraciones armónicas,
- rotaciones moleculares,
- tunelización de electrones,
- características angulares y radiales de las orbitales atómicas,
- y simetría de grupo de puntos y cómo se usa para etiquetar orbitales y vibraciones
Necesitas conocer este material si deseas entender la mayor parte de lo que ofrece este texto, por lo que te insto a que leas la Parte 1 si tu educación a la fecha aún no ha sido adecuadamente expuesta a ella.
Volvamos ahora a la discusión de cómo la teoría trata con la estructura molecular. Suponemos que conocemos la energía\(E(\{R_a\})\) en diversas localizaciones {\(R_a\)} de los núcleos. En algunos casos, denotamos esta energía\(V(R_a)\) y en otros la usamos\(E(R_a)\) porque, dentro de la aproximación Born-Oppenheimer, la energía electrónica\(E\) sirve como V potencial para los movimientos vibratorios de la molécula. Como se discutió en la Parte 1, entonces se puede realizar una búsqueda de la estructura de energía más baja (por ejemplo, encontrando dónde se desvanece el vector de gradiente\(\dfrac{∂E}{∂R_a}=0\) y donde la segunda derivada o matriz de hessian no\(\bigg(\dfrac{∂^2E}{∂R_a∂R_b}\bigg)\) tiene valores propios negativos). Al encontrar tal mínimo local en el panorama energético, la teoría es capaz de determinar una estructura estable de dicha molécula. La palabra estable se usa para describir estas estructuras no porque sean más bajas en energía que todas las demás disposiciones posibles de los átomos sino porque las curvaturas, como se dan en términos de valores propios de la matriz hessiana\(\bigg(\dfrac{∂^2E}{∂R_a∂R_a}\bigg)\), son positivas en esta geometría particular. Los procedimientos por los cuales se encuentran mínimos en el paisaje energético pueden implicar simplemente probar si la energía disminuye o aumenta a medida que cada coordenada geométrica varía en una pequeña cantidad. Alternativamente, si los gradientes\(\dfrac{∂E}{∂R_a}\) son conocidos en una geometría particular, se pueden realizar búsquedas dirigidas cuesta abajo a lo largo del negativo del propio gradiente. Al dar un pequeño paso en tal dirección, uno puede pasar a una nueva geometría que es más baja en energía. Si no solo se\(\bigg(\dfrac{∂^2E}{∂R_a∂R_a}\bigg)\) conocen los gradientes\(\dfrac{∂E}{∂R_a}\) sino también las segundas derivadas en alguna geometría, se puede dar un paso más inteligente hacia una geometría de menor energía. Para obtener detalles adicionales sobre cómo se realizan dichas búsquedas de optimización de geometría dentro del software moderno de química computacional, consulte el Capítulo 3 donde se trató este tema con mayor detalle.
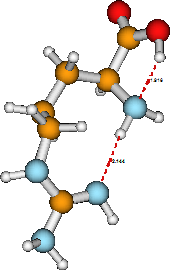
A menudo resulta que una molécula tiene más de una estructura estable (isómero) para un estado electrónico dado. Además, las geometrías que pertenecen a estructuras estables de estado electrónico excitado son diferentes a las obtenidas para el estado fundamental (porque la ocupación orbital y por lo tanto la naturaleza de la unión es diferente). Nuevamente usando arginina como ejemplo, su estado electrónico básico también tiene la estructura mostrada en la Figura 5.2 como isómero estable. Observe que este isómero y el mostrado anteriormente tienen los átomos unidos entre sí de manera idéntica, pero en la segunda estructura el grupo a-amino está involucrado en dos enlaces de hidrógeno mientras que está involucrado en solo uno en el primero. En principio, las energías relativas de estos dos isómeros geométricos se pueden determinar resolviendo la ecuación electrónica de Schrödinger mientras se colocan los núcleos constituyentes en las ubicaciones descritas en las dos figuras.
Si la molécula de arginina se excita a otro estado electrónico, por ejemplo, al promover un electrón no enlazante en su átomo de oxígeno C=O hacia el\(\pi^*\) orbital C-O vecino, sus estructuras estables no serán las mismas que en el estado electrónico básico. En particular, la distancia C-O correspondiente será más larga que en el estado del suelo, pero también se pueden modificar otros parámetros geométricos internos (aunque probablemente menos que la distancia C-O). Además, la reactividad química de este estado excitado de arginina será diferente a la del estado fundamental porque los dos estados tienen diferentes orbitales disponibles para reaccionar con reactivos atacantes.
En resumen, resolviendo la ecuación electrónica de Schrödinger en una variedad de geometrías y buscando geometrías donde el gradiente se desvanezca y la matriz de Hessian tenga todos los valores propios positivos, se pueden encontrar estructuras estables de moléculas (e iones). La ecuación de Schrödinger es un aspecto necesario de este proceso porque el movimiento de los electrones se rige por esta ecuación más que por ecuaciones clásicas newtonianas. La información obtenida después de llevar a cabo dicho proceso de optimización de geometría incluye (1) todas las distancias interatómicas y ángulos internos necesarios para especificar la geometría de equilibrio {\(R_{a{\rm eq}}\)} y (2) la energía electrónica total\(E\) en esta geometría particular.
También es posible extraer mucha más información de estos cálculos. Por ejemplo, multiplicando elementos de la matriz hessiana\(\bigg(\dfrac{∂^2E}{∂R_a∂R_b}\bigg)\) por las raíces cuadradas inversas de las masas atómicas de los átomos etiquetados a y b, se forma la hessiana ponderada en masa (\(\dfrac{1}{\sqrt{m_am_b}}\dfrac{∂^2E}{∂R_a∂R_b}\)) cuyos valores propios distintos de cero dan las frecuencias vibracionales armónicas {\(\omega_k\)} de la molécula. Los vectores propios {\(R_{k,a}\)} de la matriz Hessiana ponderada en masa dan los desplazamientos relativos en coordenadas\(R_{ka}\) que acompañan a la vibración en el modo\(k^{\rm th}\) normal (es decir, describen los movimientos del modo normal). Los detalles sobre cómo se obtienen estas frecuencias vibracionales armónicas y modos normales se discutieron anteriormente en el Capítulo 3.
Cambio molecular- reacciones e interacciones
1.Cambios en la unión
La química también se ocupa de las transformaciones de la materia, incluidos los cambios que ocurren cuando las moléculas reaccionan, se excitan (electrónicamente, vibracionalmente o rotacionalmente) o se someten a reordenamientos geométricos. Nuevamente, la teoría forma la piedra angular que permite conectar sondas experimentales de cambio químico al nivel molecular y que permite simulaciones de tales cambios.
La excitación molecular puede implicar o no alterar la estructura electrónica de la molécula; la excitación vibratoria y rotacional no, pero la excitación electrónica, la ionización y la unión electrónica sí. Como se ilustra en la Figura 5.3 donde se muestra una reacción bi-molecular, las reacciones químicas implican romper algunos enlaces y formar otros, y por lo tanto implican reordenamiento de los electrones entre diversos orbitales moleculares.
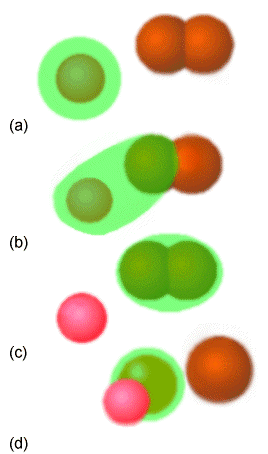
En este ejemplo, en la parte (a) el átomo verde choca con la molécula diatómica marrón y forma la molécula triatómica unida (b). Alternativamente, en (c) y (d), un átomo rosado choca con un diatómico verde para romper el enlace entre los dos átomos verdes y formar un nuevo enlace entre los átomos rosa y verde. Ambas reacciones se denominan bi-moleculares porque el paso básico en el que tiene lugar la reacción requiere una colisión entre especies independientes (es decir, el átomo y la diatómica).
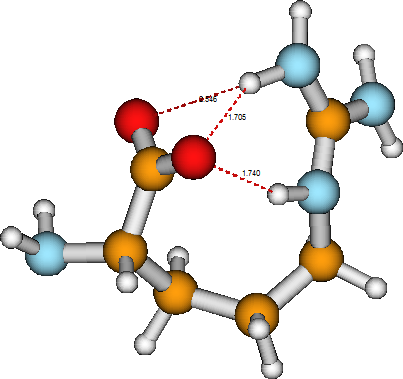
Un ejemplo sencillo de una reacción química unimolecular es ofrecido por la molécula de arginina considerada anteriormente. En la primera estructura mostrada para la arginina, el grupo ácido carboxílico conserva su\(HOOC-\) unión. Sin embargo, en la estructura zwitterión de esta misma molécula, mostrada en la Figura 5.4, el\(HOOC-\) grupo ha sido desprotonado para producir un grupo anión carboxilato\(–COO^-\), con el\(H^+\) ión ahora unido al grupo imina terminal, convirtiéndolo así en un grupo amino y colocando la carga positiva neta en el átomo de carbono adyacente. La reacción de tautomerización unimolecular en la que se interconvierten las dos formas de arginina implica romper un\(O-H\) enlace, formar un\(N-H\) enlace y cambiar un doble enlace carbono-nitrógeno en un enlace sencillo carbono-nitrógeno. En tal proceso, la estructura electrónica se altera significativamente y, como resultado, los dos isómeros pueden mostrar reactividades químicas muy diferentes hacia otros reactivos. Observe que, una vez más, la estructura última del tautómero zwitterión de la arganina está determinada por las preferencias de valencia de sus átomos constituyentes así como por los enlaces de hidrógeno formados entre diversos grupos funcionales (el grupo carboxilato y un grupo amino y un\(–NH^-\) grupo).
Conservación de Energía
En cualquier reacción química como en todos los procesos físicos (distintos del evento nuclear en el que la masa y la energía pueden ser interconvechosas), se debe conservar la energía total. Las reacciones en las que la suma de las resistencias de todos los enlaces químicos en los reactivos excede la suma de las fuerzas de unión en los productos se denominan endotérmicas. Para tales reacciones, la energía externa debe proporcionar a las moléculas reaccionantes para permitir que ocurra la reacción. Las reacciones exotérmicas son aquellas para las que los enlaces en los productos superan en fuerza a los de los reactivos. Para las reacciones exotérmicas, no se necesita un aporte neto de energía para permitir que la reacción tenga lugar. En cambio, el exceso de energía se genera y libera cuando se producen tales reacciones. En el primer caso (endotérmico), la energía que necesita la reacción suele provenir de la energía cinética de las moléculas reaccionantes o moléculas que las rodean. Es decir, la energía térmica del medio ambiente proporciona la energía necesaria. Análogamente, para las reacciones exotérmicas, el exceso de energía producido a medida que avanza la reacción se deposita generalmente en la energía cinética de las moléculas del producto y en la de las moléculas circundantes. Para reacciones que son muy endotérmicas, puede ser prácticamente imposible que la excitación térmica proporcione suficiente energía para efectuar la reacción. En tales casos, puede ser posible utilizar una fuente de luz (es decir, fotones cuya energía puede excitar las moléculas reaccionantes) para inducir la reacción. Cuando la fuente de luz provoca la excitación electrónica de los reactivos (por ejemplo, uno podría excitar un electrón en la molécula diatómica unida discutida anteriormente de un enlace a un orbital antienlace), se habla de inducir reacción por medios fotoquímicos.
Conservación de la simetría orbital: las reglas de Woodward-Hoffmann
Un ejemplo de lo importante que es entender los cambios en la unión que acompañan a una reacción química, consideremos una reacción en la que el 1,3-butadieno se convierte, mediante cierre de anillo, para formar ciclobuteno. Específicamente, enfocarse en los cuatro\(\pi\) orbitales del 1,3-butadieno a medida que la molécula sufre el llamado cierre disrotatorio a lo largo del cual se conserva el plano de simetría que biseca y es perpendicular al\(C_2-C_3\) enlace. Los orbitales del reactivo y del producto pueden ser etiquetados como pares e o impares bajo reflexión a través de este plano de simetría. No es apropiado etiquetar los orbitales con respecto a su simetría bajo el plano que contiene los cuatro átomos de C porque, aunque este plano es efectivamente una operación de simetría para los reactivos y productos, no sigue siendo una simetría válida a lo largo de la trayectoria de reacción. Es decir, al aplicar las reglas de Woodward-Hoffmann, etiquetamos simetría los orbitales usando solo aquellos elementos de simetría que se conservan a lo largo de la trayectoria de reacción que se examina.
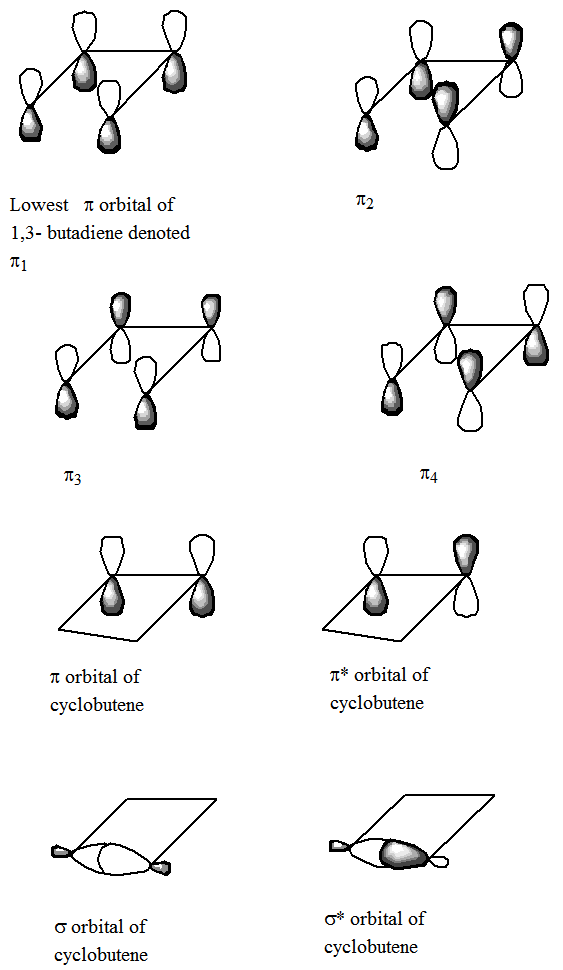
Los cuatro\(\pi\) orbitales de 1,3-butadieno son de las siguientes simetrías bajo el plano de simetría preservado (ver los orbitales en la Figura 5.5):\(\pi_1= e, \pi_2= o, \pi_3=e, \pi_4= o\). Los\(\sigma^*\) orbitales\(\pi\)\(\pi^*\)\(\sigma\) y y y del producto ciclobutano, que evolucionan a partir de los cuatro orbitales del 1,3-butadieno, son del siguiente orden de simetría y energía:\(\sigma = e, \pi = e, \pi^* = o, \sigma^* = o\). Las reglas de Woodward-Hoffmann nos instruyen a organizar los orbitales de reactivos y productos en orden de aumentar la energía y luego conectar estos orbitales por simetría, comenzando por el orbital de energía más baja y pasando por el orbital de mayor energía. Este proceso da el siguiente llamado diagrama de correlación orbital:

Luego debemos considerar cómo evolucionan las configuraciones electrónicas en las que se disponen los electrones como en el estado fundamental de los reactivos a medida que ocurre la reacción.
Notamos que los dos orbitales más bajos de los reactivos, que son los ocupados por los cuatro\(\pi\) electrones del reactivo, no se conectan a los dos orbitales más bajos de los productos, que son los orbitales ocupados por los dos\(\sigma\) y dos\(\pi\) electrones de los productos. Esto hace que la configuración de estado fundamental de los reactivos (\(\pi_1{}^2 \pi_2{}^2\)) evolucione hacia una configuración excitada (\(\sigma^2 \pi^{*2}\)) de los productos. Esto, a su vez, produce una barrera de activación para el reordenamiento térmico disrotatorio (en el que los cuatro electrones activos ocupan estos dos orbitales más bajos) de 1,3-butadieno para producir ciclobuteno.
Si los reactivos pudieran prepararse, por ejemplo, por fotólisis, en un estado excitado que tiene ocupación orbital\(\pi_1{}^2\pi_2{}^1\pi_3{}^1\), entonces la reacción a lo largo de la trayectoria considerada no tendría ninguna barrera impuesta por simetría porque esta configuración excitada individualmente se correlaciona con una configuración\(\sigma^2\pi^1\pi^{*1}\) de excitación única del productos. El hecho de que las configuraciones de reactivo y producto sean de nivel de excitación equivalente provoca que no haya restricciones de simetría en la reacción inducida fotoquímicamente de 1,3-butadieno para producir ciclobuteno. En contraste, la reacción térmica considerada primero anteriormente tiene una barrera impuesta por simetría porque la ocupación orbital se ve obligada a reorganizarse (por la ocupación de dos electrones de\(\pi_2{}^2 = \pi^{*2}\) a\(\pi^2 = \pi_3{}^2\)) para que la función de onda de estado fundamental del reactivo evolucione suavemente hacia la del producto. Por supuesto, si los reactivos pudieran generarse en un estado excitado teniendo ocupación\(\pi_1{}^2 \pi_3{}^2\) orbital, entonces los productos también podrían producirse directamente en su estado electrónico básico. Sin embargo, es difícil, si no imposible, generar tales estados electrónicos doblemente excitados, por lo que es raro que uno encuentre reacciones inducidas a través de tales estados.
Cabe destacar que aunque estas consideraciones de simetría pueden permitir anticipar barreras en las superficies de energía potencial de reacción, no tienen nada que ver con las diferencias de energía termodinámica de tales reacciones. Lo que aborda el tratamiento de simetría Woodward-Hoffmann anterior es si habrá barreras impuestas por la simetría por encima y más allá de cualquier diferencia de energía termodinámica. Las entalpías de formación de reactivos y productos contienen la información sobre el balance energético general de la reacción y deben considerarse independientemente del tipo de análisis de simetría orbital que se acaba de introducir.
Como ilustra el ejemplo anterior, es importante tener en cuenta si se produce una reacción química en el suelo o en una superficie electrónica de estado excitado. Este ejemplo muestra que uno podría querer fotoexcitar las moléculas reaccionantes para hacer que la reacción ocurra a una velocidad acelerada. Con los electrones ocupando los orbitales de menor energía, la reacción de cierre del anillo aún puede ocurrir, pero tiene que superar una barrera para hacerlo (puede emplear energía térmica de colisión para superar esta barrera), por lo que su velocidad podría ser lenta. Si se excita un electrón, no hay barrera de simetría que superar, por lo que la velocidad puede ser mayor. Las reacciones que tienen lugar en estados excitados también tienen la oportunidad de producir productos en estados electrónicos excitados, y tales productos de estado excitado pueden emitir luz. Tales reacciones se denominan quimioluminiscentes porque producen luz (luminiscencia) por medio de una reacción química.
Tasas de cambio
Las tasas de reacciones juegan un papel crucial en muchos aspectos de nuestras vidas. Las tasas de diversas reacciones biológicas determinan qué tan rápido metabolizamos los alimentos, y las tasas a las que se queman los combustibles en el aire determinan si se producirá una explosión o una llama tranquila. Los químicos ven que la velocidad de cualquier reacción entre moléculas (y tal vez fotones o electrones si se utilizan para inducir excitación en moléculas reaccionantes) está relacionada con (1) la frecuencia con la que las especies reaccionantes se encuentran entre sí y (2) la probabilidad de que un conjunto de tales especies reaccione una vez que lo hacen encontrarnos unos con otros. Los primeros aspectos se relacionan principalmente con las concentraciones de las especies reaccionantes y las velocidades con las que se mueven. Estos últimos tienen más que ver con si las especies que se encuentran colisionan en una orientación favorable (por ejemplo, ¿la enzima y el sustrato se acoplan correctamente, o el\(Br^-\) ion choca con el\(H_3C^-\) final de\(H_3C-Cl\) o con el\(Cl\) final en la reacción de S N 2 que rinde\(CH_3Br + Cl^-\)? ) y con energía suficiente para superar cualquier barrera que deba pasarse para efectuar la ruptura de los enlaces en los reactivos para formar nuevos enlaces en los productos.
Las velocidades de las reacciones pueden alterarse cambiando las concentraciones de las especies reaccionantes, cambiando la temperatura o añadiendo un catalizador. Las concentraciones y la temperatura controlan las tasas de colisión entre las moléculas, y la temperatura también controla la energía disponible para superar las barreras. Los catalizadores son moléculas que no se consumen durante la reacción pero que provocan que se incremente la velocidad de la reacción (las especies que ralentizan la velocidad de una reacción se denominan inhibidores). La mayoría de los catalizadores actúan proporcionando orbitales propios que interactúan con los orbitales de las moléculas reaccionantes para hacer que las energías de estas últimas se reduzcan a medida que avanza la reacción. En la reacción de cierre del anillo citada anteriormente, los orbitales del catalizador interactuarían (es decir, se superpondrían) con los\(\pi\) orbitales del 1,3-butadieno de una manera que disminuyera sus energías y, por lo tanto, redujera la barrera energética que debe superarse para que la reacción continúe
Además de ser capaz de determinar las geometrías (longitudes y ángulos de enlace), energías, frecuencias vibracionales de especies como los isómeros de arginina discutidos anteriormente, la teoría también aborda cuestiones de cómo y qué tan rápido ocurren las transiciones entre estos isómeros. La cuestión de cómo ocurren las reacciones químicas se centra en el mecanismo de la reacción, es decir, cómo se mueven los núcleos y cómo cambian las ocupaciones orbitales electrónicas a medida que el sistema evoluciona de reactivos a productos. En cierto sentido, comprender el mecanismo de una reacción en detalle equivale a tener una imagen mental en movimiento de cómo se mueven los átomos y electrones a medida que ocurre la reacción.
La cuestión de qué tan rápido ocurren las reacciones se relaciona con las tasas de reacciones químicas. En la mayoría de los casos, las velocidades de reacción están determinadas por la frecuencia con la que las moléculas reaccionantes acceden a una geometría crítica (llamada estado de transición o complejo activado) cerca de la cual se produce la ruptura del enlace y la formación del enlace La energía potencial de las moléculas reaccionantes a lo largo de la ruta que conecta los reactivos a través de un estado de transición para producir a menudo se representa como se muestra en la Figura 5.7.
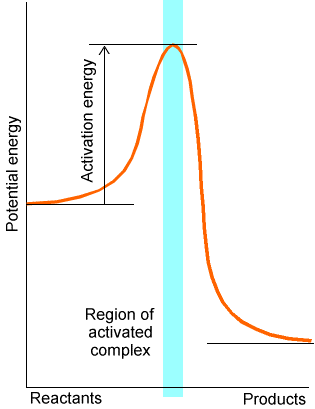
Figura 5.7 Gráfica de progreso de energía vs. reacción que muestra el estado de transición o complejo activado y la energía de activación.
En esta figura, la energía potencial (es decir, la energía electrónica sin la energía cinética de los núcleos incluida) se representa a lo largo de una coordenada que conecta los reactivos a los productos. Las geometrías y energías de los reactivos, productos y del complejo activado se pueden determinar utilizando los métodos de búsqueda de energía potencial en la superficie discutidos brevemente anteriormente y detallados anteriormente en el Capítulo 3. El capítulo 8 proporciona más información sobre la teoría de las velocidades de reacción y cómo dichas velocidades dependen de las propiedades geométricas, energéticas y vibracionales de las moléculas reaccionantes.
Las frecuencias con las que se accede al estado de transición están determinadas por la cantidad de energía (denominada energía de activación\(E^*\)) necesaria para acceder a esta geometría crítica. Para sistemas en o cerca de equilibrio térmico, la probabilidad de que la molécula gane energía\(E^*\) se muestra para tres temperaturas en la Figura 5.8.
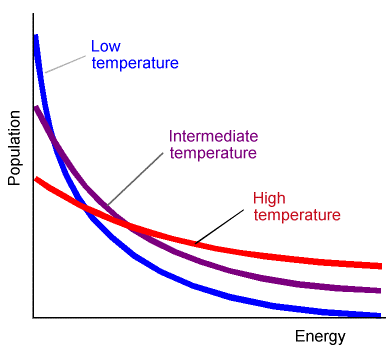
Para tales casos, las velocidades de reacción química suelen mostrar una dependencia de la temperatura caracterizada por gráficas lineales de\(\ln(k)\) vs\(1/T\). Por supuesto, no todas las reacciones involucran moléculas que han sido preparadas en o cerca del equilibrio térmico. Por ejemplo, en experimentos supersónicos de haz molecular, la distribución de energía cinética de las moléculas colisionantes es más probable que sea del tipo mostrado en la Figura 5.9.
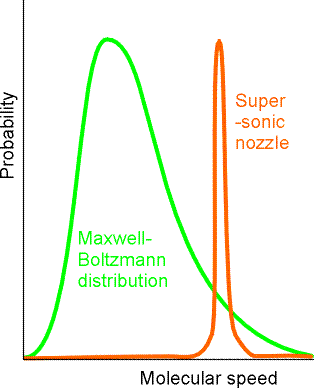
En esta figura, se grafica la probabilidad en función de la velocidad relativa con la que colisionan las moléculas reaccionantes. Es común al hacer tales gráficas de velocidad de colisión incluir el factor de elemento de\(v^2\) volumen en la parcela. Es decir, la distribución de probabilidad normalizada para que las moléculas que tienen masa reducida m colisionen con componentes de velocidad relativa\(v_z, v_y, v_z\) es
\[P(v_x, v_y, v_z) dv_x dv_y dv_z = \bigg(\dfrac{\mu}{2\pi kT}\bigg)^{3/2} \exp\bigg(-\dfrac{\mu(v_x^2+v_y^2+v_z^2)}{2kT}\bigg) dv_x dv_y dv_z.\]
Debido a que solo la energía cinética de colisión total es importante para superar las barreras de reacción, convertimos esta distribución de componentes de velocidad cartesiana en uno en términos de\(v = \sqrt{v_x^2+v_y^2+v_z^2}\) la velocidad de colisión. Esto se hace cambiando de coordenadas cartesianas a polares (en las que la variable radial es v misma) y da (después de integrarse sobre las dos coordenadas angulares):
\[P(v) dv = 4p \bigg(\dfrac{\mu}{2\pi kT}\bigg)^{3/2} \exp\bigg(-\dfrac{\mu v^2}{2kT}\bigg) v^2 dv.\]
Es el\(v^2\) factor en esta distribución de velocidad el que hace que la distribución de Maxwell-Boltzmann desaparezca a bajas velocidades en la gráfica anterior.
Otro tipo de experimento en el que se utilizan condiciones no térmicas para extraer información sobre las energías de activación ocurre dentro del ámbito de las reacciones de moléculas de iones donde se utiliza la disociación inducida por colisión (CID) para romper una molécula. Por ejemplo, cuando un complejo que consiste en un\(Na^+\) catión unido a una molécula de uracilo es acelerado por un campo eléctrico externo a una energía cinética\(E\) y posteriormente se le permite impactar en una muestra gaseosa de átomos de Xe, la colisión de alta energía permite que la energía cinética se convierta en interna energía. Esta transferencia de energía colisional puede depositar en el complejo Na+ (uracilo) suficiente energía para fragmentar el\(Na^+\)... uracilo energía de unión atractiva, produciendo\(Na^+\) así fragmentos de uracilo neutros. Si la señal para la producción de\(Na^+\) es monitoreada a medida que aumenta la energía de colisión,\(E\) se genera un perfil de velocidad de reacción CID tal como se muestra en la Figura 5.10.
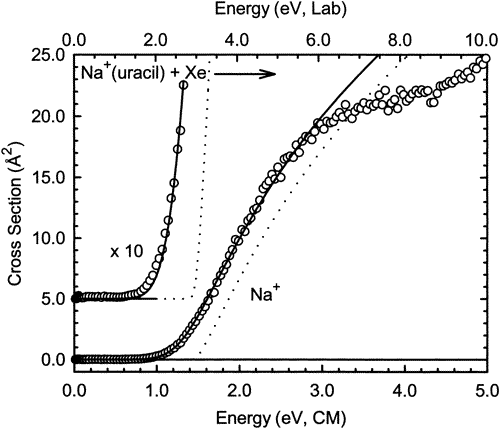
En el eje vertical se traza una cantidad proporcional a la velocidad a la que se forman los\(Na^+\) iones. En el eje horizontal se grafica la energía de colisión\(E\) en dos formatos. La energía cinética de laboratorio es simplemente 1/2 de la masa del\(Na^+({\rm uracil})\) complejo multiplicada por el cuadrado de la velocidad de estos complejos iónicos medida con respecto a un marco de coordenadas fijado en laboratorio. La energía cinética del centro de masa (CM) es la cantidad de energía disponible entre el\(Na^+({\rm uracil})\) complejo y el átomo de Xe, y viene dada por
\[E_{\rm CM} = \dfrac{1}{2} \dfrac{m_{\rm complex} m_{Xe}}{m_{\rm complex} + m_{Xe}} v^2,\]
donde\(v\) está la velocidad relativa del complejo y el átomo de Xe,\(m_{Xe}\) y\(m_{\rm complex}\) son las respectivas masas de los socios colisionantes.
La lección más esencial para aprender de dicha gráfica es que no se produce disociación si\(E\) está por debajo de algún valor umbral crítico, y la reacción CID
\[Na^+({\rm uracil}) \rightarrow Na^+ + {\rm uracil}\]
ocurre con una tasa cada vez mayor a medida que la energía de colisión\(E\) aumenta más allá del umbral. Para el ejemplo mostrado anteriormente, la energía umbral es ca. 1.2-1.4 eV. Estos umbrales CID nos pueden proporcionar estimaciones de endotermicidades de reacción y son especialmente útiles cuando estas energías exceden en gran medida de lo que se puede lograr simplemente calentando la muestra.
Mecánica Estadística: Tratamiento de Grandes Números de Moléculas en Contacto Cercano
Cuando se tiene un gran número de moléculas que sufren colisiones frecuentes (intercambiando así energía, impulso e impulso angular), el comportamiento de esta colección de moléculas a menudo se puede describir de una manera sencilla. A primera vista, parece poco probable que el tratamiento de un gran número de moléculas pueda requerir mucho menos esfuerzo que el requerido para describir una o algunas de esas moléculas.
Para ver la esencia de lo que estoy sugiriendo, considere una muestra de 10 cm 3 de agua a temperatura ambiente y presión atmosférica. En esta muestra macroscópica, hay aproximadamente 3.3 x10 23 moléculas de agua. Si uno imagina tener un instrumento que pudiera monitorear la velocidad instantánea de una molécula seleccionada, se esperaría que la señal instrumental mostrara un comportamiento irregular muy desigual si la señal fuera monitoreada en escalas de tiempo del orden del tiempo entre colisiones moleculares. En esta escala de tiempo, la molécula de agua que se está monitoreando puede estar moviéndose lentamente en un instante, pero, al colisionar con un vecino, pronto puede estar moviéndose muy rápidamente. En contraste, si se monitorea la velocidad de esta sola molécula de agua en una escala de tiempo muy larga (es decir, mucho más larga que el tiempo promedio entre colisiones), se obtiene un cuadrado promedio de la velocidad que está relacionada con la temperatura\(T\) de la muestra vía\(\dfrac{1}{2} mv^2 = \dfrac{3}{2} kT\). Esta relación se mantiene porque la muestra está en equilibrio a temperatura\(T\).
Un ejemplo del tipo de comportamiento que describo anteriormente se muestra en la Figura 5.11.
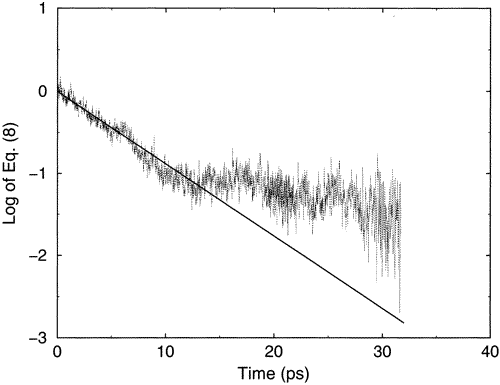
En esta figura, en el eje vertical se grafica el log de la energía (cinética más potencial) de un solo\(CN^-\) anión en una solución con agua como disolvente en función del tiempo. La etiqueta del eje vertical dice la Ec. (8) porque esta cifra fue tomada de un artículo de literatura. El\(CN^-\) ion inicialmente tiene exceso de energía vibratoria en esta simulación que se llevó a cabo en parte para modelar el flujo de energía de este ion soluto caliente a las moléculas de disolvente circundantes. Uno ve claramente los rápidos sacudidas en la energía que experimenta este ion mientras sufre colisiones con moléculas de agua vecinas. Estas sacudidas ocurren aproximadamente cada 0.01 ps, y algunas de ellas corresponden a colisiones que toman energía del ion y otras a colisiones que le dieron energía al ion. En escalas de tiempo más largas (por ejemplo, más de 1-10 ps), también vemos una caída gradual en el contenido de energía del\(CN^-\) ion que ilustra la pérdida lenta de su exceso de energía en la escala de tiempo más larga.
Ahora, consideremos qué sucede si monitoreamos un gran número de moléculas en lugar de una sola molécula dentro de la muestra de 1 cm 3 de la\(H_2O\) mencionada anteriormente. Si imaginamos dibujar una esfera de radio\(R\) y monitorear la velocidad promedio de todas las moléculas de agua dentro de esta esfera, obtenemos una imagen cualitativamente diferente si la esfera es lo suficientemente grande como para contener muchas moléculas de agua. Para R grande, se encuentra que el cuadrado promedio de la velocidad de todas las moléculas de\(N\) agua que residen dentro de la esfera (es decir,\(\displaystyle\sum_{K =1}^N \dfrac{1}{2} mv_K^2\)) es independiente del tiempo (incluso cuando se considera en una secuencia de tiempos separados por fracciones de ps) y está relacionado con la temperatura a\(T\) través\(\displaystyle\sum_K \dfrac{1}{2} mv_K^2 = \dfrac{3N}{2} kT\).
Este ejemplo muestra que, en equilibrio, el promedio a largo plazo de una propiedad de cualquier molécula individual es el mismo que el promedio instantáneo de esta misma propiedad sobre un gran número de moléculas. Para la molécula única, se logra el valor promedio de la propiedad promediando su comportamiento a lo largo de escalas de tiempo que duran muchas, muchas colisiones. Para la recolección de muchas moléculas, se logra el mismo valor promedio (en cualquier instante de tiempo) porque el número de moléculas dentro de la esfera (que es proporcional a\(\dfrac{4}{3} \pi R^3\)) es mucho mayor que el número cerca de la superficie de la esfera (proporcional a\(4\pi R^2\)) que las moléculas interiores a la esfera está esencialmente en equilibrio para todos los tiempos.
Otra forma de decir lo mismo es notar que las fluctuaciones en el contenido de energía de una sola molécula son muy grandes (es decir, la molécula sufre frecuentes grandes sacudidas) pero duran poco tiempo (es decir, el tiempo entre colisiones). En contraste, para una colección de muchas moléculas, las fluctuaciones en la energía para toda la colección son pequeñas en todo momento porque las fluctuaciones se producen por intercambio de energía con las moléculas que no están dentro de la esfera (y así se relacionan con la relación superficie a volumen de la esfera).
Entonces, si uno tiene una gran cantidad de moléculas que uno tiene razones para creer que están en equilibrio térmico, se puede evitar intentar seguir la dinámica instantánea detallada de corto tiempo de cualquier molécula o de todas las moléculas. En cambio, uno puede enfocarse en las propiedades promedio de toda la colección de moléculas. Lo que esto significa para una persona interesada en simulaciones teóricas de tales problemas de medios condensados es que no hay necesidad de realizar una simulación de dinámica molecular newtoniana del sistema (o una simulación cuántica) si está en equilibrio porque se pueden encontrar los promedios a largo plazo de lo que se calcula de otra manera. Cómo se logra esto es a través de la magia de la mecánica estadística y la termodinámica estadística. Uno de los dispositivos más potentes de la mecánica estadística es el denominado algoritmo de simulación de Montecarlo. Tales herramientas teóricas proporcionan una manera directa de calcular promedios de equilibrio (y pequeñas fluctuaciones sobre dichos promedios) para sistemas que contienen un gran número de moléculas. En el Capítulo 7, proporciono una breve introducción a los fundamentos de esta subdisciplina de la química teórica donde aprenderás más sobre este apasionante campo.
A veces hablamos del comportamiento de equilibrio o del comportamiento dinámico de una colección de moléculas. Permítanme elaborar un poco sobre lo que significan estas frases. Las propiedades de equilibrio de las colecciones moleculares incluyen las funciones de distribución radial y angular entre diversos centros atómicos. Por ejemplo, las funciones O-O y de distribución\(O-H\) radial en agua líquida y en hielo se muestran en la Figura 5.12.
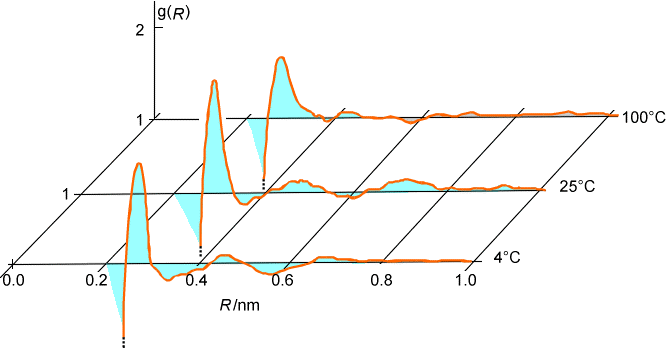
Dichas propiedades representan promedios, a lo largo de largos tiempos o sobre una gran colección de moléculas, de alguna propiedad que no cambia con el tiempo excepto en una escala de tiempo muy rápida correspondiente a colisiones individuales.
Por el contrario, las propiedades dinámicas de las colecciones moleculares incluyen los procesos de plegamiento y despliegue que experimentan las proteínas y otros polímeros; las migraciones de protones de molécula de agua a molécula de agua en agua líquida y a lo largo de\(H_2O\) cadenas dentro de canales iónicos; y el autoensamblaje de moléculas monocapas sobre superficies sólidas a medida que varía la concentración de las moléculas en la capa superior líquida. Estas son propiedades que ocurren en escalas de tiempo mucho más largas que aquellas entre colisiones moleculares y en escalas de tiempo que deseamos sondear por algún experimento o por simulación.
Habiendo introducido brevemente las áreas primarias de la química teórica: estructura, dinámica y mecánica estadística, examinemos ahora cada una de ellas con algo más detalle, teniendo en cuenta que los Capítulos 6-8 son donde cada uno es tratado con mayor detalle.
Colaboradores y Atribuciones
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry
Integrated by Tomoyuki Hayashi (UC Davis)