
1.3: imágenes
- Page ID
- 76844
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)1. Apuntes de conferencia
2. Química Física IV
3. Parte 2: Electron
Gunnar Jeschke
25
20

4. Derechos de autor (c) 2016 Gunnar Jeschke
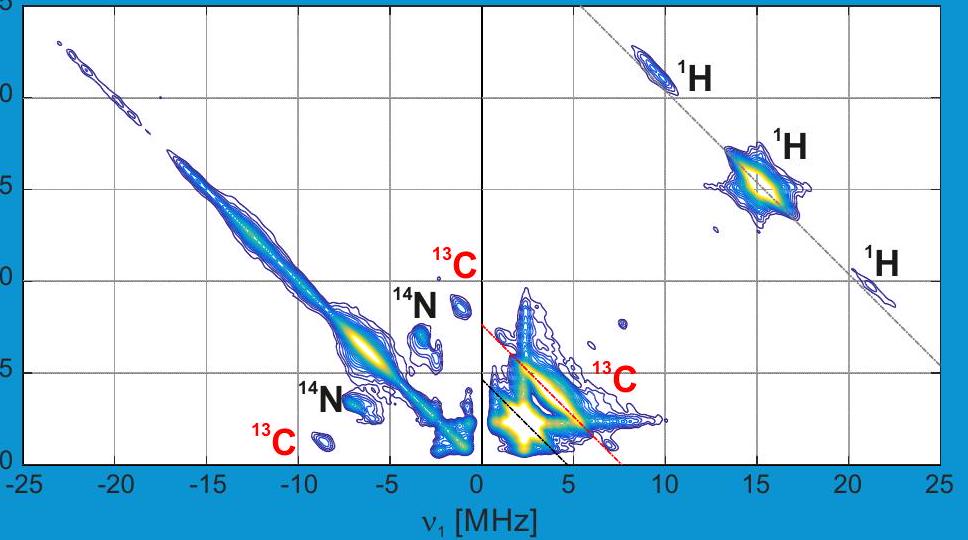
Imagen del título: Espectro HYSCORE de una especie superficial de Ti (III) (en colaboración con C. Copéret, F. Allouche, V. Kalendra)
Capítulo 2 Placa de memoria Stern-Gerlach: Pluma (Licencia GNU Free Documentation)
Capítulo 3 Efecto Zeeman sobre líneas espectrales (fotografía de Pieter Zeeman, 1897)
Capítulo 4 Interacción hiperfina por polarización de espín (obra propia)
Capítulo 5 Interacción dipolo-dipolo (obra propia)
Capítulo 6 Esquema de nivel de energía de un sistema de centrifugado (obra propia)
Capítulo 7 Modulación de campo en CW EPR (obra propia)
Capítulo 8 Traza ESEEM de deuterio de una proteína marcada con espín (obra propia)
Capítulo 9 Medición de la distribución de distancia en un constructo de ARN marcado con espín (colaboración con O. Duss, M. Yulikov, F. H.-T. Allain)
Capítulo 10 Estructura electrónica y marco molecular de una etiqueta giratoria de nitróxido (obra propia)
5. PUBLICADO POR GUNNAR JESCHKE
http://WWW. epr. ethz. ch
Licenciado bajo la Licencia Creative Commons Reconocimiento-NoComercial Unported (la “Licencia”). No podrá utilizar este archivo salvo en cumplimiento de la Licencia. Puede obtener una copia de la Licencia en http://creativecommons. org/licencias/by-nc/3.0.
El diseño y la disposición de las notas de la conferencia se basan en el Libro Naranja de Legrand disponible en http://latextemplates. com/template/the-legrand-orange e-book.
Primera impresión, Octubre 2016
6. Contenidos
Introducción
espín de electrones
Resonancia magnética del electrón libre
2.1.1 El momento magnético del electrón libre
2.2 Interacciones en sistemas de espín electrón-nuclear 10
2.2.1 Consideración general sobre las interacciones de espín
2.2.2 El giro electrón-nuclear Hamiltoniano.... 12
Interacción Zeeman de 3 electrones... 15
3.1 Origen físico del turno 15
3.2 Electron Zeeman Hamiltoniano 17
3.3 Manifestación espectral de la interacción del electrón Zeeman 18
3.3.1 Solución líquida
Estado sólido
Interacción hiperfina
4.1 Origen físico de la interacción hiperfina 21
Interacción de contacto de Fermi
Polarización de giro... 23
4.2 Hamiltoniano hiperfino 24 4.3 Manifestación espectral de la interacción hiperfina 25
4.3.2 Espectros de frecuencia nuclear de solución líquida
Espectros EPR de estado sólido... 28
Espectros de frecuencia nuclear de estado sólido
5 Interacciones Electrón-Electrón...
5.1 Interacción de intercambio 31
Intercambio hamiltoniano... 32
5.1.3 Manifestación espectral de la interacción de intercambio....
5.2 Interacción dipolo-dipolo 33
Imagen física
Dipolo-dipolo Hamiltoniano
Manifestación espectral de la interacción dipolo-dipolo............ 35
5.3.1 Imagen física

6 Transiciones Electrón-Nucleares Prohibidas.... 43
6.1 Imagen física 43
6.1.1 El
Campos locales en el giro nuclear... 43
6.2 Formalismo operador de producto con interacciones pseudo-seculares 45
Transformación de a la propia base
6.2.2 Cálculos generales de operador de producto para un Hamiltoniano no diagonal... 46
6.3 Generación y detección de coherencia nuclear por excitación de espín electrónico 47
Generador de coherencia nuclear
Espectroscopia EPR de 7 CW... 49
7.1 Por qué y cómo se realiza la espectroscopia CW EPR 49
7.1.1 Ventajas de sensibilidad de la espectroscopia CW EPR...
7.1.3 Consideraciones sobre la preparación de muestras
7.2 Descripción teórica de CW EPR 53
8 Medición de Acoplamientos Hiperfinos Pequeños...... 57
8.1 ENDOR 57
8.1.1 Ventajas de la detección de espectros de frecuencia nuclear basada en espín electrónico
8.1.2 Tipos de experimentos ENDOR... 57
8.1.3 Davies ENDOR 8.2 ESEEM e HYSCORE
8.2.1 ¿ENDOR o ESEEM?
8.2.2 ESEEM de tres pulsos
HYSCORE
Mediciones de distribución de distancia
9.1.1 El experimento DEER de cuatro pulsos
9.1.2 Requerimientos de muestra
9.2.1 Expresión para la señal DEER
Corrección de fondo
9.2.3 Regularización Tikhonov con restricción de no negatividad.... 72
Sondas de centrifugado y trampas de giro
10.1.1 Sondas giratorias y etiquetas
Radicales Nitróxido
El espectro de EPR de Nitróxido
Influencia de la dinámica en el espectro del nitróxido
Polaridad y proticidad
Accesibilidad al agua
Accesibilidad al oxígeno
Mediciones locales
Trampas giratorias 83

Libros 85
Artículos 85
Índice


7. 1 - Introducción
7.1. Observaciones Generales
La espectroscopia de Resonancia Paramagnética Electrónica (EPR) es menos conocida y menos aplicada que la espectroscopia de RMN. La razón es que la espectroscopia EPR requiere electrones desapareados y el emparejamiento electrónico suele ser energéticamente favorable. De ahí que solo una pequeña fracción de sustancias puras exhiba señales EPR, mientras que la espectroscopia de RMN es aplicable a casi cualquier compuesto que se pueda imaginar. Por otro lado, como el emparejamiento de electrones subyace al enlace químico, los electrones desapareados están asociados con la reactividad. En consecuencia, la espectroscopia EPR es una técnica muy importante para comprender las reacciones radicales, los procesos de transferencia de electrones y la catálisis de metales de transición, todos ellos relacionados con la 'reactividad del electrón desapareado'. Algunas especies con electrones desapareados son químicamente estables y pueden ser utilizadas como sondas de espín para estudiar sistemas donde la espectroscopia de RMN se encuentra en límites de resolución o no puede proporcionar información suficiente para la caracterización completa de la estructura y la dinámica. Este curso presenta los conceptos básicos para aplicar espectroscopía EPR en especies reactivas o catalíticamente activas, así como en sondas de espín.
Muchos conceptos en espectroscopia EPR están relacionados con conceptos similares en espectroscopía de RMN. Por lo tanto, las conferencias sobre espectroscopia EPR se basan en material que se ha introducido anteriormente en las conferencias sobre espectroscopia de RMN. Este material se repite brevemente y se realza en este guion y se señalan similitudes así como diferencias. Tal tratamiento vinculado de las dos técnicas no se encuentra en los libros de texto introductorios. Al enfatizar este vínculo, el curso enfatiza la comprensión de la física que subyace a la espectroscopia de RMN y EPR en lugar de enfocarse en campos de aplicación individuales. Nuestro objetivo es comprender los espectros a nivel fundamental y comprender cómo se pueden medir los parámetros del giro hamiltoniano con la mejor sensibilidad y resolución posibles.
El capítulo 2 del guión introduce el espín electrónico, lo relaciona con el espín nuclear y discute, qué interacciones contribuyen al giro hamiltoniano de un sistema paramagnético. El capítulo 3 trata la interacción del electrón Zeeman, la desviación del valor de un electrón unido del valor de un electrón libre, y la manifestación de anisotropía en espectros EPR de estado sólido. El capítulo 4 introduce la interacción hiperfina entre espines electrónicos y nucleares, que proporciona la mayor parte de la información sobre la estructura electrónica y espacial de los centros paramagnéticos. La manifestación espectral en estado líquido y sólido se considera para los espectros del espín electrónico y de los espines nucleares. El capítulo 5 discute los fenómenos que ocurren cuando la interacción hiperfina es tan grande que se viola la aproximación de campo alto para el espín nuclear. En esta situación, las transiciones formalmente prohibidas se permiten parcialmente y la mezcla de los niveles de energía conduce a cambios en las frecuencias de resonancia. El capítulo 6 discute cómo se describe el acoplamiento entre espines de electrones en el giro hamiltoniano, dependiendo de su tamaño. A lo largo de los Capítulos, las interacciones introducidas del espín electrónico están relacionadas con la estructura electrónica y espacial.
Los capítulos 7-9 están dedicados a las técnicas experimentales. En el Capítulo 7, se introduce la EPR de onda continua como la técnica más versátil y sensible para medir espectros de EPR. Los requisitos para obtener espectros bien resueltos con alta relación señal/ruido se derivan de los primeros principios físicos. En el capítulo 8 se analizan dos técnicas para medir acoplamientos hiperfinos en espectros de frecuencia nuclear, donde se resuelven mejor que en los espectros EPR. Los experimentos de doble resonancia nuclear de electrones (ENDOR) utilizan polarización de espín electrónico y detección de espines electrónicos para mejorar la sensibilidad de tales mediciones, pero aún dependen de la excitación directa de los espines nucleares. Los experimentos de modulación de envolvente de eco de espín electrónico (ESEEM) se basan en las transiciones de espín electrón-nuclear prohibidas discutidas en el Capítulo El capítulo 9 trata la medición de las distribuciones de distancia en el rango de nanómetros separando el acoplamiento dipolo-dipolo entre espines de electrones de otras interacciones.
El Capítulo 10 final introduce el sondeo de espín y el atrapamiento de espín y, al mismo tiempo, demuestra la aplicación de conceptos que se introdujeron en Capítulos anteriores.
En algunos puntos (acoplamiento dipolo-dipolo, explicación de la espectroscopia CW EPR en términos de las ecuaciones de Bloch) este guión de conferencia se superpone significativamente con la parte de RMN del guión de la conferencia. Esto se pretende con el fin de hacer que el script EPR sea razonablemente autónomo. Obsérvese también que este guión de conferencia tiene dos propósitos. En primer lugar, debe servir de ayuda en el estudio de la materia y la preparación para el examen. En segundo lugar, es material de referencia cuando posteriormente encuentras especies paramagnéticas en tu propia investigación y necesitas obtener información sobre ellas por espectroscopia EPR.
7.2. Lectura sugerida y recursos electrónicos
No existe ningún libro de texto sobre espectroscopia EPR que trate todo el material de este curso en un nivel básico. Sin embargo, muchos de los conceptos están cubiertos por un título de la serie Oxford Chemistry Primer de Chechik, Carter y Murphy [CCM16]. Los estudiantes con mentalidad física también pueden apreciar el libro de texto estándar más antiguo de Weil, Bolton y Wertz [WBW94].
Para algunos de los espectros simulados y ejemplos trabajados en estas notas de conferencia, se proporcionan scripts de Matlab o cuadernos de Mathematica en la página principal de la conferencia. Parte de las simulaciones numéricas se basa en EasySpin de Stefan Stoll (http://wWW. easyspin.org/) y otra parte sobre SPIDYAN de Stephan Pribitzer (http://www. epr. ethz. ch/software. html). Los cálculos con formalismo de operador de producto requieren el paquete Mathematica Spinop.m de Serge Boentges, que está disponible en la página principal del curso. Un paquete alternativo más grande para tales cálculos analíticos es SpinDynamica de Malcolm Levitt (http://Www. spindynamica. soton. ac. uk/). Por último, pero no menos importante el paquete más extenso para simulaciones numéricas de experimentos de resonancia magnética es SPINACH de Ilya Kuprov et al. (http://spindynamics. org/ Spinach.php). Para los cálculos químico-cuánticos de los parámetros hamiltonianos de espín, el programa probablemente más versátil es el paquete ORCA disponible gratuitamente (https://orcaforum. cec.mpg.de/).
8. Resonancia magnética del electrón libre
El momento magnético del electrón libre Diferencias entre la espectroscopia EPR y RMN
Interacciones en sistemas de espín electrón-nuclear Consideración general sobre las interacciones de espín El espín electrón-nuclear Hamiltoniano

9. 2 - Espín de electrones


10. Resonancia magnética del electrón libre
10.2.1. El momento magnético del electrón libre
Como partícula elemental, el electrón tiene un momento angular intrínseco llamado espín. El número cuántico de espín es, de manera que en un campo magnético externo a lo largo, solo se pueden observar dos valores posibles para el componente de este momento angular, correspondientes al estado del número cuántico magnético y, correspondiente al número cuántico magnético (estado. La diferencia de energía entre los dos estados correspondientes del electrón resulta del momento magnético asociado con el giro. Para una partícula giratoria clásica con carga elemental, momento angular y masa, este momento magnético calcula para
La relación carga-masa es mucho mayor para el electrón que la relación correspondiente para un núcleo, donde es del orden de, donde está la masa protónica. Al introducir el magnetón Bohr y el factor de corrección cuántico-mecánica, podemos reescribir la ecuación (2.1) como
La mecánica cuántica Dirac relativista proporciona, una corrección que también se puede encontrar en una derivación no relativista. Las mediciones exactas han demostrado que el valor de un electrón libre se desvía ligeramente de. La corrección necesaria se puede derivar por electrodinámica cuántica, conduciendo a. La diferencia de energía entre los dos estados de espín de un electrón libre en un campo magnético externo viene dada por
de manera que la relación giromagnética del electrón libre es. Esta relación giromagnética corresponde a una frecuencia de resonancia de a un campo de, que es por un factor de aproximadamente 658 mayor que la frecuencia nuclear Zeeman de un protón.
10.2.2. Diferencias entre EPR y espectroscopía de RMN
La mayoría de las diferencias entre la espectroscopia RMN y EPR son el resultado de este momento magnético mucho mayor del electrón. La polarización de Boltzmann es mayor por este factor y al mismo campo magnético los fotones detectados tienen una energía mayor por este factor. Los tiempos de relajación son aproximadamente por un factor más cortos, lo que permite una repetición mucho más rápida de experimentos de EPR en comparación con los experimentos de RMN Como resultado, la espectroscopia EPR es mucho más sensible. La instrumentación estándar con un electroimán que trabaja en un campo de aproximadamente y a frecuencias de microondas de aproximadamente (banda X) puede detectar alrededor de espines, si la muestra tiene pérdidas dieléctricas insignificantes por microondas. En solución acuosa, los radicales orgánicos pueden detectarse en concentraciones menores a en un tiempo de medición de pocos minutos.
Debido al gran momento magnético del espín electrónico, la aproximación de alta temperatura puede ser violada sin usar equipos exóticos. La energía de transición de espín de un electrón libre coincide con la energía térmica a una temperatura de y un campo de aproximadamente correspondiente a una frecuencia de aproximadamente (banda W). Asimismo, la aproximación de campo alto puede descomponerse. La interacción dipolo-dipolo entre dos espines de electrones es por un factor de mayor que entre dos protones y dos electrones desapareados pueden acercarse entre sí que dos protones. La división de campo cero que resulta de dicho acoplamiento puede ascender a una fracción significativa de la interacción de Zeeman de electrones o incluso puede excederla en los campos magnéticos, donde generalmente se realizan experimentos de EPR. El acoplamiento hiperfino entre un electrón y un núcleo puede superar fácilmente la frecuencia nuclear de Zeeman, lo que conduce a la ruptura de la aproximación de campo alto para el espín nuclear.
11. Interacciones en sistemas de espín electrón-nuclear
11.2.3. Consideración general sobre las interacciones de giro
Los giros interactúan con campos magnéticos. La interacción con un campo magnético externo estático es la interacción Zeeman, que suele ser la interacción de espín más grande. En campos suficientemente grandes, donde se mantiene la aproximación de campo alto, la interacción Zeeman determina la dirección de cuantificación del giro. En esta situación, es un buen número cuántico y, si la aproximación de campo alto también se mantiene para un giro nuclear, el número cuántico magnético también es un buen número cuántico. Las energías de todos los niveles de espín pueden ser expresadas por parámetros que cuantifican las interacciones de espín y por los números cuánticos magnéticos. El vector de todos los números cuánticos magnéticos define el estado del sistema de espín.
Los giros también interactúan con los campos magnéticos locales inducidos por otros giros. Por lo general, los electrones desapareados son raros, por lo que cada espín electrónico interactúa con varios espines nucleares en sus proximidades, mientras que cada espín nuclear interactúa con un solo espín de electrones (Fig. 2.1). La interacción hiperfina entre el electrón y el espín nuclear suele ser mucho menor que la interacción Zeeman del electrón, con excepciones para los iones de metales de transición. En contraste, para núcleos cercanos al espín electrónico, la interacción hiperfina puede ser mayor que la interacción nuclear de Zeeman en los campos donde generalmente se miden los espectros EPR. En este caso, que se discute en el Capítulo 6, la aproximación de campo alto se descompone y no es un buen número cuántico. Los acoplamientos hiperfinos a los núcleos son relevantes siempre que sean al menos tan grandes como la tasa de relajación transversal del espín nuclear acoplado. Los acoplamientos más pequeños no están resueltos.
En algunos sistemas, dos o más electrones desapareados están tan cerca uno del otro que su acoplamiento excede sus velocidades de relajación transversal. De hecho, la parte isotrópica de este acoplamiento puede superar con mucho la interacción de Zeeman de electrones y a menudo incluso la energía térmica si dos electrones desapareados residen en diferentes orbitales moleculares de la misma molécula orgánica (molécula de estado triplete) o si varios electrones desapareados pertenecen a un estado de alto espín de un metal de transición o iones de metales de tierras raras. En esta situación, el sistema se describe mejor en una representación acoplada con un

Figura 2.1: Esquema de interacciones en sistemas de espín electrón-nuclear. Todos los giros tienen una interacción Zeeman con el campo magnético externo. Los espines de electrones (rojos) interactúan entre sí por la interacción dipolo-dipolo a través del espacio y por intercambio debido a la superposición de los orbitales moleculares ocupados individualmente (verde). Cada espín electrónico interactúa con espines nucleares (azules) en su vecindad por acoplamientos hiperfinos (púrpura). Los acoplamientos entre espines nucleares suelen ser insignificantes en los sistemas paramagnéticos, al igual que los desplazamientos químicos. Estas dos interacciones son demasiado pequeñas en comparación con la tasa de relajación en las proximidades de un espín de electrones.
espín del grupo de electrones. El acoplamiento isotrópico entre los espines de electrones individuales no influye en la división del subnivel para un número cuántico de espín de grupo dado. El acoplamiento anisotrópico, que conduce a la división de subniveles, se denomina interacción de campo cero o fina. Si la interacción de Zeeman de electrones excede con mucho el acoplamiento espín-espín, es más conveniente describir el sistema en términos de los espines electrónicos individuales. El acoplamiento de intercambio isotrópico, que proviene del solapamiento de dos orbitales moleculares ocupados individualmente (SOM), contribuye entonces a la división de niveles. Además, el acoplamiento dipolo-dipolo a través del espacio entre dos espines electrónicos también contribuye.
Concepto - Orbital molecular de ocupación individual (SOMO). Cada orbital molecular puede estar ocupado por dos electrones con un número cuántico de espín magnético opuesto. Si un orbital molecular está ocupado individualmente, el electrón no está apareado y su número cuántico de espín magnético se puede cambiar por absorción o emisión de fotones. El orbital ocupado por el electrón desapareado se denomina orbital molecular de ocupación individual (SOMO). Varios electrones desapareados pueden existir en la misma molécula o complejo metálico, es decir, puede haber varios SOM.
Los espines nucleares en las proximidades de un espín de electrones se relajan mucho más rápido que los espines nucleares en sustancias diamagnéticas. Sus tasas de relajación transversal superan así los acoplamientos entre los espines nucleares y los desplazamientos químicos. Estas interacciones, que son muy importantes en la espectroscopia de RMN, son insignificantes en la espectroscopia EPR. Para espines nucleares no se puede obtener información sobre la identidad química de un núcleo, a menos que se entienda su acoplamiento hiperfino. El elemento puede ser identificado a través de la interacción nuclear Zeeman. Para los espines nucleares, la información sobre la identidad química se codifica en la interacción cuadrupolo nuclear, cuya magnitud generalmente excede.
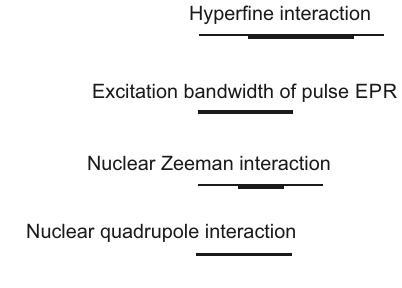
En la Figura 2.2 se ofrece una visión general de todas las interacciones y su magnitud típica en unidades de frecuencia. Esta Figura también ilustra otra diferencia entre la espectroscopía EPR y RMN. Varias interacciones, como la interacción de campo cero, la interacción hiperfina, los acoplamientos dipolo-dipolo más grandes y de intercambio entre espines de electrones y también la anisotropía de la interacción Zeeman de electrones generalmente exceden el ancho de banda de excitación de los pulsos de microondas más fuertes y más cortos
Hay una excepción. Si la tasa de relajación longitudinal del espín electrónico excede con diferencia la interacción nuclear de Zeeman, la relajación del espín nuclear apenas se ve afectada por la presencia del espín electrónico. En esta situación, la espectroscopia EPR es imposible, sin embargo. que están disponibles. Las secuencias de pulsos de RMN que dependen de la capacidad de excitar el espectro completo de un cierto tipo de espines, por lo tanto, no se pueden adaptar fácilmente a la espectroscopia EPR.
11.2.4. El giro electrón-nuclear Hamiltoniano
Considerando todas las interacciones discutidas en la Sección 2.2.1, el giro estático hamiltoniano de un sistema de espín electrón-nuclear en unidades de frecuencia angular puede escribirse como
donde el índice se extiende sobre todos los espines nucleares, los índices y los espines de electrones y el símbolo denota la transposición de un vector u operador vectorial. A menudo, solo hay que considerar a la vez un espín de electrones y un espín nuclear, de modo que el giro hamiltoniano simplifica drásticamente. Para los espines de grupo de electrones, los términos con mayores potencias de los operadores de giro pueden ser significativos. No consideramos esta complicación aquí.
La interacción del electrón Zeeman es, en general, anisotrópica y por lo tanto parametrizada por tensores. Se discute en detalle en el Capítulo 3. En la interacción nuclear de Zeeman, las frecuencias nucleares de Zeeman dependen únicamente del elemento y el isótopo y así pueden especificarse sin conocer la estructura electrónica y espacial de la molécula. La interacción hiperfina vuelve a ser anisotrópica y, por lo tanto, se caracteriza por tensores. Se discute en detalle en el Capítulo 4. Todas las interacciones electrón-electrón se explican en el Capítulo 5. La interacción de campo cero es puramente anisotrópica y, por lo tanto, se caracteriza por tensores sin trazas. La interacción de intercambio es a menudo puramente isotrópica y cualquier contribución anisotrópica no puede distinguirse experimentalmente de la interacción dipolo-dipolo puramente anisotrópica. De ahí que la primera interacción se caracteriza por escalares y la segunda interacción por tensores. Finalmente, la interacción cuadrupolo nuclear se caracteriza por tensores sin rastro.

Interacción Electron Zeeman
Interacción de campo cero
Interacción dipolo-dipolo
entre espines electrónicos débilmente acoplados
Anchos de línea EPR homogéneos

Figura 2.2: Magnitud relativa de las interacciones que contribuyen al Hamiltoniano de los sistemas de espín electrón-nuclear.

Origen físico del desplazamiento Electron Zeeman Hamiltoniano Manifestación espectral de la interacción Zeeman del electrón
Solución líquida Estado sólido
12. 3 - Interacción Zeeman Electron
13. Origen físico del turno
Se encuentra que los electrones unidos tienen valores que difieren del valor para el electrón libre. Dependen de la orientación del centro paramagnético con respecto al vector de campo magnético. La razón principal de este cambio de valor es el acoplamiento del espín al momento angular orbital del electrón. El acoplamiento espín-órbita es un efecto puramente relativista y, por lo tanto, es mayor si los orbitales de átomos pesados contribuyen al SOMO. En la mayoría de las moléculas, el momento angular orbital se apaga en el estado fundamental. Por esta razón, el SOC solo conduce a cambios pequeños o moderados y puede tratarse como una perturbación. Tal tratamiento de perturbación no es válido si el estado fundamental es degenerado o casi degenerado.
El tratamiento de perturbación considera estados excitados donde el electrón desapareado no está en el SOMO del estado fundamental. Dichos estados excitados se mezclan ligeramente con el estado fundamental y la mezcla surge del operador de momento angular orbital. Por simplicidad, consideramos un caso donde la principal contribución al cambio surge de orbitales localizados en un solo átomo dominante y por SOC de un solo electrón. A segundo orden en la teoría de la perturbación, los elementos de la matriz del tensor se pueden expresar como
donde es un delta de Kronecker, el factor en el término de desplazamiento es la constante de acoplamiento espín-órbita para el átomo dominante, y los elementos de la matriz se calculan como
donde los índices y atropellan las direcciones cartesianas, y. Los operadores, y son componentes cartesianos del operador de momento angular, designa el orbital donde reside el electrón desapareado en una configuración electrónica de estado excitado, contado desde para el SOMO de la configuración de estado fundamental. La energía de ese orbital es.
Dado que el producto de las integrales superpuestas en el numerador en el lado derecho de la ecuación (3.2) suele ser positivo, el signo del desplazamiento está determinado por el denominador. El denominador es positivo si un electrón emparejado de un orbital completamente ocupado se promueve al SOMO en estado fundamental y negativo si el electrón desapareado se promueve a un orbital previamente desocupado (Figura Estado fundamental
Excitación de un electrón emparejado

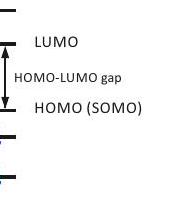
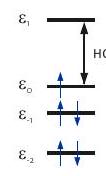

Figura 3.1: La mezcla de estados excitados por operadores de momento angular orbital conduce a un desplazamiento por acoplamiento espín-órbita. La diferencia de energía en la expresión de perturbación es positiva para la excitación de un electrón emparejado al SOMO en estado fundamental y negativa para la excitación del electrón emparejado a un orbital de mayor energía.
3.1). Debido a que la brecha de energía entre el SOMO y la órbita desocupada más baja (LUMO) suele ser mayor que la que existe entre los orbitales ocupados, los términos con numerador positivo dominan en la suma en el lado derecho de la ecuación (3.2). Por lo tanto, los cambios positivos se encuentran con mayor frecuencia que los negativos.
La constante de acoplamiento espín-órbita relevante depende del elemento y tipo de órbita. Escala aproximadamente con, donde esta la carga nuclear. A menos que exista un estado excitado muy bajo (cerca de la degeneración del estado fundamental), predominan las contribuciones de núcleos pesados. Si no son ninguno, como en los radicales orgánicos que consisten únicamente en hidrógeno y elementos de segunda fila, se observan desplazamientos de solo, los desplazamientos típicos son. Tenga en cuenta que esto aún excede los cambios químicos típicos en RMN en una a dos órdenes de magnitud. Para los metales de transición de primera fila, los cambios son del orden de.
Para los iones de tierras raras, el tratamiento de perturbación se descompone. El factor Landé se puede calcular a partir del símbolo de término para un doblete de niveles
donde es el número cuántico para el momento angular total y el número cuántico para el momento angular orbital. Los valores principales del tensor son, y, donde los con son diferencias entre los valores propios de para los dos niveles.
Si se conoce la estructura de un centro paramagnético, el tensor puede ser calculado por química cuántica. Esto funciona bastante bien para los radicales orgánicos y razonablemente bien para la mayoría de los iones de metales de transición de primera fila. Los detalles se explican en [KBE04].
El tensor es una propiedad global del SOMO y es fácilmente interpretable solo si está dominado por la contribución en un solo átomo, lo que suele ser, pero no siempre, el caso de los complejos de metales de transición y iones de tierras raras. Si el centro paramagnético tiene un eje de simetría con, el tensor tiene simetría axial con valores principales. Para simetría cúbica o tetraédrica el valor es isotrópico, pero no necesariamente igual a. También se encuentran valores isotrópicos para una muy buena aproximación para iones de metales de transición y metales de tierras raras con conchas semillenas, como en los complejos Mn (II) (configuración electrónica) y complejos Gd (III).
14. Electron Zeeman Hamiltoniano
Consideramos un solo espín electrónico y así bajamos la suma y el índice en la Ec. (2.4). En el sistema de ejes principales (PAS) del tensor, podemos entonces expresar el electrón Zeeman Hamiltoniano como
donde está el campo magnético,, y son los valores principales del tensor y los ángulos polares y determinan la orientación del campo magnético en el PAS.
Este hamiltoniano está diagonalizado por la transformación de Bleaney, proporcionando
con el valor efectivo en la orientación
Si la anisotropía del tensor es significativa, el eje en la ecuación (3.5) se inclina desde la dirección del campo magnético. Este efecto es insignificante para la mayoría de los radicales orgánicos, pero no para los iones de metales de transición o iones de tierras raras. La ecuación (3.6) para los valores efectivos describe un elipsoide (Figura).

Figura 3.2: Elipsoide que describe la dependencia de orientación del valor efectivo en el PAS del tensor. En una dirección dada del vector de campo magnético (rojo), corresponde a la distancia entre el origen y el punto donde interseca la superficie elipsoide.
Concepto 3.2.1 - Niveles de energía en la aproximación de campo alto. En la aproximación de campo alto la contribución de energía de un término hamiltoniano al nivel con números cuánticos magnéticos y puede calcularse reemplazando a los operadores por los números cuánticos magnéticos correspondientes. Esto se debe a que los números cuánticos magnéticos son los valores propios de los operadores, todos los operadores viajan entre sí, y las contribuciones con todos los demás operadores de giro cartesianos son insignificantes en esta aproximación. Para el electrón Zeeman contribución de energía es. Si la aproximación de campo alto es ligeramente violada, esta expresión corresponde a un tratamiento de perturbación de primer orden. La regla de selección para las transiciones en la espectroscopia EPR es y se aplica estrictamente siempre y cuando la aproximación de campo alto se aplique estrictamente a todos los giros. Esta regla de selección es el resultado de la conservación del momento angular en la absorción de un fotón de microondas y del hecho de que el fotón de microondas interactúa con las transiciones de espín electrónico. De ello se deduce que la contribución de primer orden de la interacción de Zeeman de electrones a las frecuencias de todas las transiciones de espín electrónico es la misma, a saber. Como veremos en el Capítulo 7, los espectros EPR generalmente se miden a frecuencia constante de microondas barriendo el campo magnético. El campo de resonancia es entonces dado por
Para las transiciones de espín nuclear,, la interacción del electrón Zeeman no contribuye a la frecuencia de transición.
14.1. Manifestación espectral de la interacción del electrón Zeeman
14.1.1. Solución líquida
En solución líquida, las moléculas se tambalean debido a la difusión rotacional browniana. La escala de tiempo de este movimiento puede caracterizarse por un tiempo de correlación rotacional que en disolventes no viscosos es del orden de para moléculas pequeñas, y del orden de hasta 100 ns para proteínas y otras macromoléculas. Para una molécula globular con radio en un disolvente con viscosidad, el tiempo de correlación rotacional puede estimarse aproximadamente por la ley Stokes-Einstein
Si este tiempo de correlación y la diferencia máxima entre las frecuencias de transición de dos orientaciones cualesquiera de la molécula en el campo magnético cumplen la relación, la anisotropía se promedia completamente y solo se observa el promedio isotrópico de las frecuencias de transición. Para una rotación algo más lenta, la modulación de la frecuencia de transición por volteo molecular conduce al ensanchamiento de la línea ya que acorta el tiempo de relajación transversal. En el régimen de caída lenta, donde la anisotropía se promedia de manera incompleta y el ancho de línea alcanza un máximo. Para, se observa el espectro de estado sólido. Los fenómenos pueden describirse como un intercambio multisitio entre las diversas orientaciones de la molécula (ver Sección 10.1.4), lo cual es análogo al intercambio químico discutido en la parte de RMN del curso de conferencia.
Para la interacción de Zeeman de electrones, el volteo rápido conduce a un campo de resonancia promedio
con el valor isotrópico. Para pequeños radicales orgánicos en disolventes no viscosos a frecuencias de banda X alrededor, el ensanchamiento de la línea a partir de la anisotropía es insignificante. A frecuencias de banda W de radicales orgánicos y ya a frecuencias de banda X para pequeños complejos de metales de transición, dicho ensanchamiento puede ser sustancial. Para macromoléculas grandes o en solventes viscosos, se pueden observar espectros de estado sólido como EPR en solución líquida.
14.1.2. Estado sólido
Para una muestra monocristalina, el campo de resonancia en cualquier orientación dada puede calcularse mediante la ecuación (3.7). A menudo, solo están disponibles polvos microcristalinos o la muestra se mide en solución congelada vítrea. En tales condiciones, todas las orientaciones contribuyen por igual. Con respecto a la

Figura 3.3: Forma de línea de polvo para un tensor con simetría axial. (a) La densidad de probabilidad para encontrar una orientación con ángulo polar es proporcional a la circunferencia de un círculo un ángulo en una esfera unitaria. b) Densidad de probabilidad. El valor efectivo en ángulo es. (c) Forma esquemática de la línea de polvo. El patrón corresponde a para un barrido de campo y a para un barrido de frecuencia. Debido a la inclinación del marco, el valor isotrópico no se encuentra en el ángulo mágico, aunque el desplazamiento es pequeño si.
ángulos polares, esto implica que se distribuye uniformemente, mientras que la probabilidad de encontrar un cierto ángulo es proporcional a (Figura 3.3). La forma de la línea del espectro de absorción se entiende más fácilmente para la simetría axial del tensor. Las transiciones se observan solo en el rango entre los campos de resonancia limitantes en y. El espectro tiene un máximo global en y un mínimo en.
En la espectroscopia CW EPR no observamos la forma de la línea de absorción, sino su primera derivada (ver Capítulo 7). Esta forma de línea derivada tiene características nítidas en las singularidades de forma de línea del espectro de absorción y amplitud muy débil en el medio (Figura).
Concepto 3.3.1 - Selección de orientación. La dispersión del espectro de una muestra de polvo o solución congelada vítrea permite seleccionar moléculas con una cierta orientación con respecto al campo magnético. Para un tensor axial solo se seleccionan orientaciones cercanas al eje del tensor PAS al observar cerca del campo de resonancia de. En contraste, al observar cerca del campo de resonancia para, las orientaciones dentro de todo el plano del PAS contribuyen. Para el caso de simetría ortorrómbica con tres valores principales distintos, y, se pueden observar conjuntos estrechos de orientaciones en los campos de resonancia correspondientes a los valores extremos y (ver panel superior derecho en la Figura 3.4). En el valor principal intermedio contribuye un amplio rango de orientaciones, ya que el mismo campo de resonancia puede ser realizado por orientaciones distintas a y. Dicha selección de orientación puede mejorar la resolución de los espectros ENDOR y ESEEM (Capítulo 8) y simplificar su interpretación.

ortorrómbico
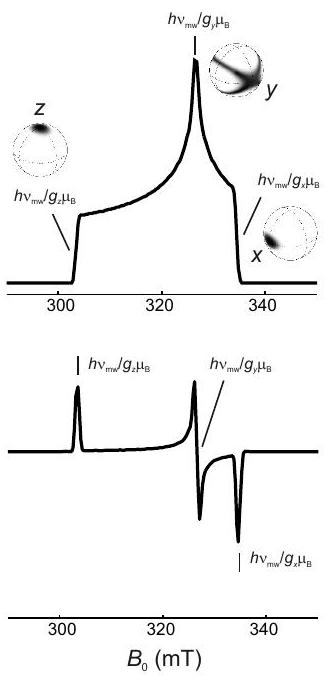
Figura 3.4: Espectros de EPR de banda X simulados para sistemas con solo anisotropía. Los paneles superiores muestran espectros de absorción, ya que se pueden medir mediante espectroscopía EPR de barrido de campo con detección de eco. Los paneles inferiores muestran la primera derivada de los espectros de absorción ya que son detectados por EPR de onda continua. Las imágenes unidad-esfera en el panel superior derecho visualizan las orientaciones que se seleccionan en los campos de resonancia correspondientes a los valores principales del tensor.

14.2. Origen físico de la interacción hiperfina
Los momentos magnéticos de un electrón y un par de espín nuclear por la interacción dipolo-dipolo magnético; similar a la interacción dipolo-dipolo entre espines nucleares discutida en la parte de RMN del curso de conferencias. La principal diferencia con el caso de RMN es que, en muchos casos, una descripción punto-dipolo no es una buena aproximación para el espín electrónico, ya que el electrón se distribuye sobre el SOMO. El núcleo considerado puede ser considerado como bien localizado en el espacio. Ahora imaginamos al SOMO como una combinación lineal de orbitales atómicos. Las contribuciones de la densidad de espín en un orbital atómico de otro núcleo (población del electrón desapareado en tal orbital atómico) se pueden aproximar asumiendo que el electrón desapareado es un dipolo puntual localizado en este otro núcleo.
Para la densidad de espín en orbitales atómicos sobre un mismo núcleo, tenemos que distinguir entre tipos de orbitales atómicos. En los orbitales, el electrón desapareado tiene densidad de probabilidad finita para residir en el núcleo, a una distancia cero del espín nuclear. Esto conduce a una singularidad de la interacción dipolo-dipolo, ya que esta interacción escala con. La singularidad ha sido tratada por Fermi. La contribución al acoplamiento hiperfino a partir de la densidad de espín en orbitales sobre el núcleo bajo consideración se denomina interacción de contacto de Fermi. Debido a la simetría esférica de los orbitales, la interacción de contacto de Fermi es puramente isotrópica.
Para la densidad de espín en otros orbitales (orbitales) en el núcleo bajo consideración, la interacción dipolo-dipolo debe promediarse sobre la distribución espacial del espín electrónico en estos orbitales. Este promedio no tiene aporte isotrópico. Por lo tanto, la densidad de espín en orbitales no influye en los espectros de radicales de volteo rápido o complejos metálicos en solución líquida y tampoco la densidad de espín en orbitales de otros núcleos. Los acoplamientos isotrópicos detectados en solución resultan únicamente de la interacción de contacto de Fermi.
Dado que las contribuciones isotrópicas y puramente anisotrópicas al acoplamiento hiperfino tienen un origen físico diferente, separamos estas contribuciones en el tensor hiperfino que describe la interacción entre espín electrónico y espín nuclear:
donde está el acoplamiento isotrópico hiperfino y el acoplamiento puramente anisotrópico. En lo siguiente, bajamos los índices de espín electrónico y nuclear y.
14.2.1. Interacción hiperfina dipolo-dipolo
El tensor de acoplamiento hiperfino anisotrópico de un núcleo dado se puede calcular a partir de la función de onda del estado fundamental aplicando el principio de correspondencia a la interacción clásica entre dos dipolos puntuales
Dichos cálculos se implementan en programas de química cuántica como ORCA, ADF o Gaussiana. Si el SOMO es considerado como una combinación lineal de orbitales atómicos, las contribuciones de un orbital individual pueden expresarse como el producto de la densidad de espín en este orbital con un factor espacial que puede calcularse de una vez por todas. Los factores espaciales han sido tabulados [KM85]. En general, los núcleos de elementos con mayor electronegatividad tienen factores espaciales más grandes. Al mismo factor espacial, como para los isótopos del mismo elemento, el acoplamiento hiperfino es proporcional al valor nuclear y por lo tanto proporcional a la relación giromagnética del núcleo. Por lo tanto, un acoplamiento de deuterio puede calcularse a partir de un acoplamiento de protones conocido o viceversa.
Una situación especial se aplica a los protones, metales alcalinos y metales alcalinotérreos, que no tienen densidades de espín significativas en, o -orbitales. En este caso, la contribución anisotrópica solo puede surgir del acoplamiento dipolo-dipolo a través del espacio a centros de densidad de espín en otros núcleos. En una aproximación punto-dipolo, el tensor hiperfino viene dado por
donde la suma recorre todos los núcleos con densidad de espín significativa (sumada en todos los orbitales de este núcleo) distintos del núcleo considerado. Son distancias entre el núcleo considerado y los centros de densidad de espín, y los vectores son unidades a lo largo de la dirección desde el núcleo considerado hasta el centro de densidad de espín. Para los protones en complejos de metales de transición a menudo es una buena aproximación considerar la densidad de espín solo en el ion metálico central. La distancia desde el protón al ion central puede entonces inferirse directamente de la parte anisotrópica del acoplamiento hiperfino.
Las contribuciones de tensor hiperfino calculadas por cualquiera de estas formas deben ser corregidas por la influencia de si el tensor es fuertemente anisotrópico. Si la contribución dominante a surge en un solo núcleo, el tensor hiperfino en este núcleo puede corregirse mediante
El producto g puede tener una parte isotrópica, aunque es puramente anisotrópico. Esta contribución de pseudocontacto isotrópico depende de la orientación relativa del tensor y del tensor hiperfino dipolo-dipolo giratorio-dipolo. La corrección es insignificante para la mayoría de los radicales orgánicos, pero no para los iones metálicos paramagnéticos. Si surgen contribuciones de varios centros, la corrección necesaria no puede escribirse en función del tensor.
14.2.2. Interacción de contacto de Fermi
El aporte de contacto de Fermi toma la forma
La mayor parte de la literatura sostiene que la corrección debe hacerse para todos los núcleos. Como señaló Frank Neese, esto no es cierto. Una discusión anterior de este punto se encuentra en [Lef67] donde se encuentra la densidad de espín en el orbital bajo consideración, el valor nuclear y el magnetón nuclear. El factor denota la probabilidad de encontrar el electrón en este núcleo en el estado fundamental con función de onda y ha sido tabulado [KM85].
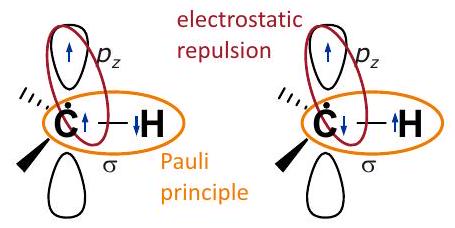
Figura 4.1: Transferencia de densidad de espín por el mecanismo de polarización de espín. Según el principio Pauli, los dos electrones en el orbital del enlace C-H deben tener un estado de espín opuesto. Si el electrón desapareado reside en un orbital en el átomo, para otros electrones en el mismo átomo se favorece ligeramente el mismo estado de espín, ya que esto minimiza la repulsión electrostática. De ahí que para el electrón en el átomo, el estado de espín opuesto (panel izquierdo) se ve ligeramente favorecido sobre el mismo estado de espín (panel derecho). La densidad de espín positiva en la órbita en el átomo induce cierta densidad de espín negativa en el orbital en el átomo.
14.2.3. Polarización de giro
Las contribuciones al acoplamiento hiperfino discutidas hasta este punto pueden ser entendidas y calculadas en una imagen de un solo electrón. Otras contribuciones surgen de la correlación de electrones en una molécula. Supongamos que el orbital sobre un átomo de carbono contribuye al SOMO, de manera que se prefiere el estado de espín del electrón en ese orbital (Fig. 4.1). Los electrones en otros orbitales del mismo átomo tendrán entonces también una ligera preferencia por el estado (panel izquierdo), ya que los electrones con el mismo giro tienden a evitarse entre sí y por lo tanto tienen menos repulsión electrostática. En particular, esto significa que la configuración de giro en el panel izquierdo de la Fig. Es ligeramente más preferible que la del panel derecho. Según el principio Pauli, los dos electrones que comparten el enlace orbital del enlace deben tener espín antiparalelo. Así, el electrón en el orbital del átomo de hidrógeno que está unido al átomo de carbono portador del espín tiene una ligera preferencia por el estado. Esto corresponde a un acoplamiento hiperfino isotrópico negativo del protón directamente unido, el cual es inducido por el acoplamiento hiperfino positivo del átomo de carbono adyacente. El efecto se denomina “polarización de espín”, aunque no tiene relación física con la polarización de las transiciones de espín electrónico en un campo magnético externo.
La polarización de espín es importante, ya que transfiere la densidad de espín desde orbitales, donde es invisible en solución líquida y de átomos de carbono con baja abundancia natural del isótopo magnético a orbitales sobre protones, donde se puede observar fácilmente en solución líquida. Esta transferencia ocurre, tanto, en radicales, donde el electrón desapareado se localiza en un solo átomo, como en radicales, donde se distribuye sobre el sistema. Este último caso es de mayor interés, ya que la distribución de la órbita sobre los núcleos puede mapearse midiendo y asignando los acoplamientos isotrópicos de protones hiperfinos. Este acoplamiento se puede predecir mediante la ecuación de McConnell
donde es la densidad de espín en el átomo de carbono adyacente y es un parámetro del orden de, que depende ligeramente de la estructura del sistema.
Esta preferencia por que los electrones del mismo átomo tengan espín paralelo es también la base de la regla de Hund.

Figura 4.2: Mapeo del LUMO y HOMO de una molécula aromática mediante mediciones de acoplamientos hiperfinos después de reducción u oxidación de un electrón. La reducción conduce a un radical anión, cuyo SOMO es una buena aproximación al orbital molecular desocupado más bajo (LUMO) de la molécula progenitora neutra. La oxidación conduce a un radical catiónico, cuyo SOMO es una buena aproximación al orbital molecular más alto ocupado (HOMO) de la molécula progenitora neutra.
La ecuación de McConnell se aplica principalmente para mapear el LUMO y HOMO de moléculas aromáticas (Figura 4.2). Un electrón desapareado puede ser puesto en estos orbitales por reducción u oxidación de un electrón, respectivamente, sin perturbar demasiado fuertemente los orbitales. Los acoplamientos isotrópicos hiperfinos del átomo de hidrógeno directamente unidos a un átomo de carbono informan sobre la contribución de la órbita de este átomo de carbono a la órbita. Los retos en este mapeo son dos. Primero, es difícil asignar los acoplamientos observados a los átomos de hidrógeno a menos que ya se disponga de un modelo para la distribución de la órbita. Segundo, el método es ciego a los átomos de carbono sin un átomo de hidrógeno unido directamente.
14.3. Hamiltoniano hiperfino
Consideramos la interacción de un solo espín electrónico con un solo espín nuclear y así caer las sumas e índices y en la Ec. (2.4). En general, todos los elementos de la matriz del tensor hiperfino serán distintos de cero después de la transformación de Bleaney al marco donde la interacción del electrón Zeeman está a lo largo del eje (ver Eq. 3.5). El hamiltoniano hiperfino es entonces dado por
Obsérvese que el eje del sistema de coordenadas de espín nuclear es paralelo al vector de campo magnético mientras que el del sistema de espín electrónico está inclinado, si la anisotropía es significativa. De ahí que el tensor hiperfino no sea un tensor en el estricto sentido matemático, sino una matriz de interacción.
En la Ec. (4.7), el término es laico y debe mantenerse siempre. Por lo general, la aproximación de campo alto sí se mantiene para el espín de electrones, de modo que todos los términos que contienen u operadores no son seculares y pueden descartarse. El hamiltoniano hiperfino truncado así dice
Los dos primeros términos en el lado derecho pueden considerarse como definiendo un acoplamiento transversal efectivo que es la suma de un vector con longitud a lo largo y un vector de longitud a lo largo. La longitud del vector suma es. El Hamiltoniano hiperfino truncado simplifica si tomamos el eje del marco de laboratorio para el giro nuclear a lo largo de la dirección de este acoplamiento hiperfino transversal efectivo. En este marco tenemos
donde cuantifica el acoplamiento hiperfino secular y el acoplamiento hiperfino pseudo-secular. Este último acoplamiento debe considerarse si y sólo si el acoplamiento hiperfino viola la aproximación de campo alto para el espín nuclear (ver Capítulo 6).
Si la anisotropía es muy pequeña, como es el caso de los radicales orgánicos, los ejes de los dos sistemas de coordenadas de espín son paralelos. En esta situación y para un tensor hiperfino con simetría axial, y puede expresarse como
donde es el ángulo entre el campo magnético estático y el eje de simetría del tensor hiperfino y es la anisotropía del acoplamiento hiperfino. Los valores principales del tensor hiperfino son y. La contribución seudo-secular se desvanece a lo largo de los ejes principales del tensor hiperfino, donde es o o para un acoplamiento hiperfino puramente isotrópico. De ahí que la contribución seudo-secular también se pueda disminuir al considerar radicales de volteo rápido en estado líquido. Ahora consideramos la aproximación punto-dipolo, donde el espín electrónico está bien localizado en la escala de longitud de la distancia electrón-nuclear y suponemos que surge únicamente de las interacciones a través del espacio. Esto se aplica a los iones hidrógeno, álcalis y alcalino-térreos. Luego encontramos
Por el momento suponemos que la contribución pseudo-secular es despreciable o puede considerarse como una pequeña perturbación. El otro caso se trata en el Capítulo 6. A primer orden, la contribución de la interacción hiperfina a los niveles de energía es dada entonces por. En el espectro EPR, cada núcleo con espín genera transiciones de espín electrónico con las que pueden ser marcadas por los valores de. En el espectro de frecuencias nucleares, cada núcleo exhibe transiciones con. Para los espines nucleares en estado sólido, cada transición se divide aún más en transiciones por la interacción cuadrupolo nuclear. La contribución del acoplamiento hiperfino secular a las frecuencias de transición electrónica es, mientras que es para las frecuencias de transición nuclear. En ambos casos, la división entre líneas adyacentes de un multiplete hiperfino viene dada por.
14.4. Manifestación espectral de la interacción hiperfina
14.4.1. Espectros EPR de solución líquida
Dado que cada núcleo divide cada transición de espín electrónico en transiciones con diferentes frecuencias, el número de transiciones EPR es. Algunas de estas transiciones pueden coincidir si los acoplamientos hiperfinos son iguales o múltiplos enteros entre sí. Un caso importante, donde los acoplamientos hiperfinos son exactamente iguales son núcleos químicamente equivalentes. Por ejemplo, dos núcleos pueden tener combinaciones de estado de espín, y. Las contribuciones a las frecuencias de transición son, y
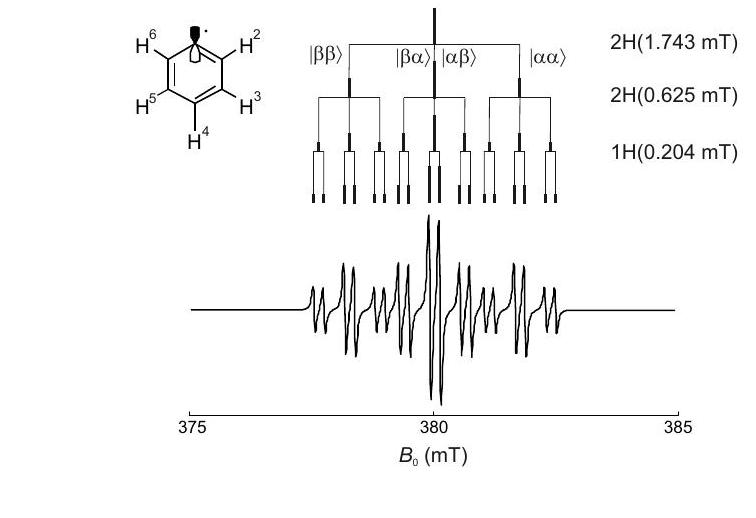
Figura 4.3: División hiperfina en el espectro EPR del radical fenilo. El mayor acoplamiento hiperfino para los dos protones orto equivalentes genera un triplete de líneas con intensidades relativas. El acoplamiento medio a los dos meta protones equivalentes divide cada línea nuevamente en un patrón, conduciendo a 9 líneas con una relación de intensidad de. Finalmente, cada línea se divide en doblete por el pequeño acoplamiento hiperfino del protón para, lo que lleva a 18 líneas con relación de intensidad.
. Para núcleos equivalentes con solo tres líneas se observan con desplazamientos hiperfinos de, y con respecto al electrón de frecuencia Zeeman. La línea central no desplazada tiene el doble de amplitud que las líneas desplazadas, lo que lleva a un patrón con división. Para núcleos equivalentes con el número de líneas es y las intensidades relativas se pueden inferir del triángulo de Pascal. Para un grupo de núcleos equivalentes con número cuántico de espín arbitrario el número de líneas es. Las multiplicidades de grupos de núcleos equivalentes se multiplican. Por lo tanto, el número total de líneas EPR es
donde el índice se extiende sobre los grupos de núcleos equivalentes.
La figura ilustra en el ejemplo del radical fenilo cómo surge el patrón multiplete. Para los radicales con sistemas más extendidos, el número de líneas puede ser muy grande y puede llegar a ser imposible resolver completamente el espectro. Incluso si el espectro está completamente resuelto, el análisis del patrón multiplete puede ser una tarea formidable. Un algoritmo que funciona bien para el análisis de patrones con un número moderado de líneas se da en [CCM16].
14.4.2. Espectros de frecuencia nuclear en solución líquida
Como se menciona en la Sección, el acoplamiento hiperfino secular se puede inferir de los espectros de frecuencia nuclear así como de los espectros de EPR. Los anchos de línea son menores en los espectros de frecuencia nuclear, ya que los espines nucleares tienen tiempos de relajación transversal más largos. Otra ventaja de los espectros de frecuencia nuclear surge del hecho de que el espín electrónico interactúa con todos los espines nucleares mientras que cada espín nuclear interactúa con un solo espín de electrones (Figura 4.4). El número de líneas en los espectros de frecuencia nuclear crece así solo linealmente con el número de núcleos, mientras que
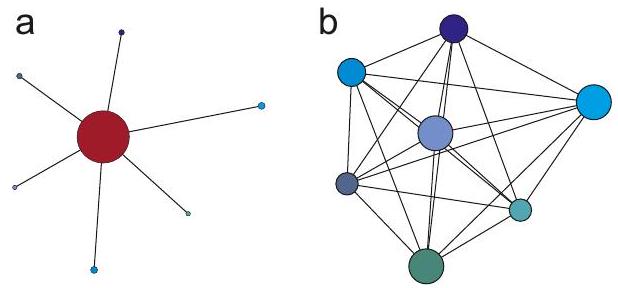
Figura 4.4: Topologías de un sistema de espín electrón-nuclear para espectroscopia EPR (a) y de un sistema de espín nuclear típico para espectroscopía de RMN (b). Debido al momento magnético mucho mayor del espín electrónico, el espín electrónico “ve” todos los núcleos, mientras que cada espín nuclear en el caso EPR ve solo el espín electrónico. En el caso de RMN, cada espín nuclear se ve espín nuclear, dando lugar a información muy rica, pero más difícil de analizar.
crece exponencialmente en espectros EPR. En solución líquida, cada grupo de espines nucleares equivalentes agrega líneas, de manera que el número de líneas para dichos grupos es
Los espectros de frecuencia nuclear en solución líquida pueden medirse mediante CW ENDOR, técnica que se discute brevemente en la Sección 8.1.2.
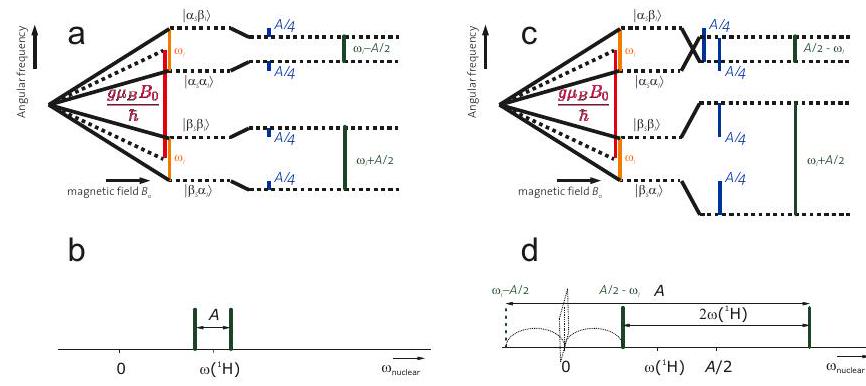

Figura 4.5: Esquemas de nivel de energía (a, c) y espectros de frecuencia nuclear (b, d) en los casos de acoplamiento hiperfino débil y acoplamiento hiperfino fuerte (c, d) para un sistema de espín electrón-nuclear,. Aquí, se supone que es negativo y se supone que es positivo. (a) En el caso de acoplamiento débil,, las dos transiciones de espín nuclear (verde) tienen frecuencias. b) En el caso de acoplamiento débil, el doblete se centra en frecuencia y se divide por. (c) En el caso de acoplamiento fuerte,, los niveles cruzan para uno de los estados de espín electrónico. Las dos transiciones de espín nuclear (verde) tienen frecuencias. d) En el caso de acoplamiento fuerte, el doblete se centra en frecuencia y se divide por.
Una complicación en la interpretación de los espectros de frecuencia nuclear puede surgir del hecho de que la interacción hiperfina puede ser mayor que la interacción nuclear de Zeeman. Esto se ilustra en la Figura 4.5. Solo en el caso de acoplamiento débil con el doblete hiperfino en los espectros de frecuencia nuclear se centra en y se divide por. En el caso de acoplamiento fuerte, los subniveles hiperfinos se cruzan para uno de los estados de espín electrónico y la frecuencia nuclear se vuelve negativa. Como no se detecta el signo de la frecuencia, la línea se encuentra en la frecuencia en su lugar, es decir, se “refleja” a la frecuencia cero. Esto da como resultado un doblete centrado en la frecuencia y dividido por. El reconocimiento de tales casos en espectros de estado líquido bien resueltos se simplifica por el hecho de que la frecuencia nuclear de Zeeman solo puede asumir unos pocos valores que se conocen si se conocen los isótopos nucleares en la molécula y el campo magnético. La figura ilustra cómo se construye el espectro de frecuencia nuclear del radical fenilo en base a tales consideraciones. El espectro tiene solo 6 líneas, en comparación con las 18 líneas que surgen en el espectro EPR en la Figura 4.3.
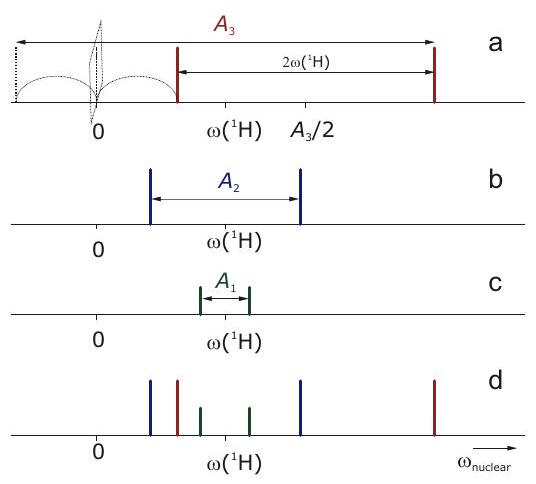
Figura 4.6: Espectro ENDOR esquemático (frecuencia nuclear) del radical fenilo a una frecuencia de banda X donde. a) Subespectro de los dos protones orto equivalentes. Se aplica el estuche de acoplamiento fuerte. b) Subespectro de los dos metaprotonesequivalentes. Se aplica el caso de acoplamiento débil. c) Subespectro del para protón. Se aplica el caso de acoplamiento débil. d) Espectro completo.
14.4.3. Espectros EPR de estado sólido
En estado sólido, la construcción de los espectros EPR se complica por el hecho de que la interacción del electrón Zeeman es anisotrópica. En cada orientación individual de la molécula, el espectro se parece al patrón en estado líquido, pero tanto la frecuencia central del multiplete como las divisiones hiperfinas dependen de la orientación. Como estas distribuciones de frecuencia son continuas, las divisiones resueltas generalmente se observan solo en las singularidades del patrón de forma de línea de la interacción con la anisotropía más grande. Para los radicales orgánicos a frecuencias de banda X, a menudo domina la anisotropía hiperfina. A altas frecuencias o para iones de metales de transición, a menudo domina la anisotropía de Zeeman de electrones. La forma exacta de la línea depende no sólo de los valores principales del tensor y de los tensores hiperfinos, sino también de la orientación relativa de sus PASs. El caso general es complicado y requiere simulaciones numéricas, por ejemplo, por EasySpin.
Sin embargo, con bastante frecuencia se encuentran casos simples, donde domina la interacción hiperfina de un solo núcleo y coinciden los PASs del tensor hiperfino. Por ejemplo, los complejos de Cu (II) suelen ser planos cuadrados y, si los cuatro ligandos son iguales, tienen un eje de simetría. El tensor que tiene simetría axial con el eje siendo el eje único. Los tensores hiperfinos de y tienen la misma simetría y el mismo eje único. Los dos isótopos tienen relaciones giromagnéticas y de espín muy similares. Los espectros se pueden entender considerando un espín electrónico y un espín nuclear con tensores axiales e hiperfinos con un eje único coincidente.
En esta situación, los subespectros para cada uno de los estados de espín nuclear, y adquieren una forma similar como se muestra en la Figura. El campo de resonancia se puede calcular resolviendo
donde está el ángulo entre el eje de simetría y el vector de campo magnético. Las singularidades se encuentran en y corresponden a frecuencias angulares y.
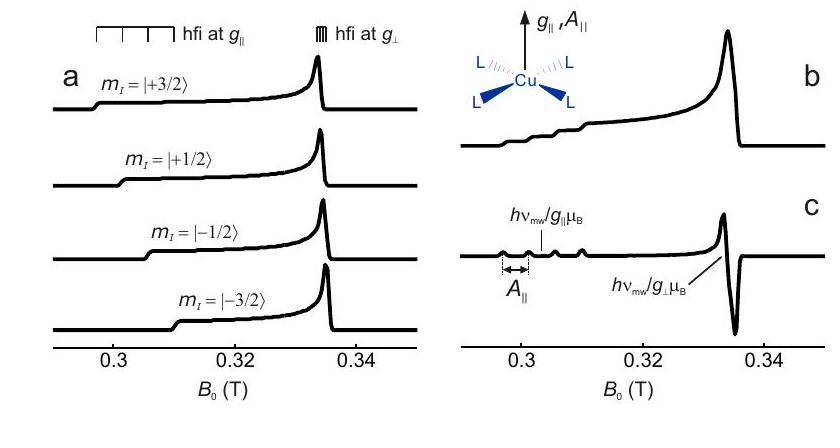
Figura 4.7: Construcción de un espectro EPR en estado sólido para un complejo de cobre (II) con cuatro ligandos equivalentes y coordinación plana cuadrada. Las direcciones de los ejes y principales coinciden con el eje de simetría del complejo (recuadro). (a) Subespectros para los cuatro estados de espín nuclear con diferente número cuántico de espín magnético. b) Espectro de absorción. (c) Derivada del espectro de absorción.
La construcción de un espectro de Cu (II) EPR de acuerdo con estas consideraciones se muestra en la Figura 4.7. Los valores de y se pueden inferir analizando las singularidades cercanas al borde de campo bajo del espectro. Cerca del borde de campo alto, la división hiperfina generalmente no se resuelve. Aquí, corresponde al máximo del espectro de absorción y al cruce por cero de su derivada.
14.4.4. Espectros de frecuencia nuclear de estado sólido
Nuevamente, se encuentra una situación más simple en los espectros de frecuencia nuclear, ya que la frecuencia nuclear de Zeeman es isotrópica y la anisotropía de desplazamiento químico es despreciablemente pequeña en comparación con la anisotropía hiperfina. Además, la resolución es mucho mejor por las razones discutidas anteriormente, de manera que se pueden detectar acoplamientos hiperfinos más pequeños y anisotropías. Si la anisotropía del acoplamiento hiperfino está dominada por el acoplamiento dipolo-dipolo a través del espacio a un solo centro de densidad de espín, como suele ser el caso de los protones, o por la contribución de la densidad de espín en un solo u orbital, como suele ser el caso de otros núcleos, el tensor hiperfino tiene simetría casi axial. En este caso, se puede inferir de las formas de línea si se aplica el caso de acoplamiento débil o fuerte y si el acoplamiento hiperfino isotrópico es positivo o negativo (Figura 4.8). El caso con corresponde al patrón de Pake discutido en la parte de RMN del curso de conferencia.
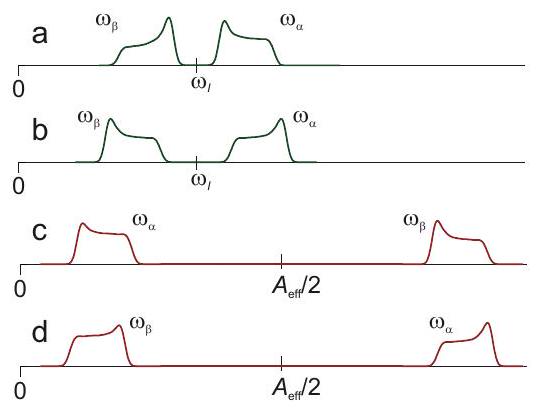
Figura 4.8: Espectros de frecuencia nuclear de estado sólido para casos con frecuencia nuclear negativa de Zeeman. a) Caso de acoplamiento débil con y. b) Caso de acoplamiento débil con y. (a) Caja de acoplamiento fuerte con y. (b) Caja de acoplamiento fuerte con y.

14.5. Interacción de intercambio
14.5.1. Origen físico y consecuencias de la interacción de intercambio
Si dos electrones desapareados ocupan SOM en la misma molécula o en moléculas espacialmente cercanas, las funciones de onda y de los dos SOM pueden superponerse. Los dos electrones desapareados pueden acoplarse a un estado singlete o a un estado triplete. La diferencia de energía entre el estado singlete y triplete es la integral de intercambio
Existen diferentes convenciones para el signo de y el factor 2 puede faltar en partes de la literatura. Con la convención de signos utilizada aquí, el estado singlete es menor en energía para positivo. Dado que el estado singlete con función de onda de giro es antisimétrico con respecto al intercambio de los dos electrones y los electrones son Fermiones, corresponde a la situación en la que los dos electrones también podrían ocupar el mismo orbital. Se trata de una superposición orbital de unión, correspondiente a un ordenamiento de espín antiferromagnético. Negativo corresponde a un estado triplete inferior, es decir, superposición orbital antiunión y ordenamiento de espín ferromagnético. El estado triplete tiene tres subestados con funciones de onda para el estado, para el estado y para el estado. El y estado son autoestados tanto en ausencia como en presencia del acoplamiento. Los estados y son autoestados para, donde está la diferencia entre las frecuencias de Zeeman de electrones de los dos espines. Para el caso contrario de, los autoestados son y. Este último caso corresponde a la aproximación de campo alto con respecto a la interacción de intercambio.
Para un intercambio fuerte,, las energías son aproximadamente para el estado singlete y y para los subestados triplete, y, respectivamente, donde está la interacción de Zeeman del electrón, que es la misma para ambos espines dentro de esta aproximación. Si, los fotones de microondas con energía no pueden excitar las transiciones entre el subespacio singlete y triplete del espacio Hilbert de espín. Entonces es conveniente usar una representación acoplada y considerar los dos subespacios separados uno del otro. El subespacio singlete corresponde a una molécula diamagnética y no contribuye a los espectros EPR. El subespacio triplete puede ser descrito por un giro grupal de los dos electrones desapareados. En la representación acoplada, no entra en el giro hamiltoniano, ya que desplaza todos los niveles subespaciales por la misma energía. Porque, el estado triplete es el estado fundamental y siempre es observable por espectroscopia EPR. Sin embargo, generalmente uno tiene y el estado singlete es el estado fundamental. Siempre y cuando no supere la energía térmica por un factor grande, el estado triplete es térmicamente excitado y observable. En este caso, la amplitud de la señal EPR puede aumentar en lugar de disminuir con el aumento de la temperatura. Para las moléculas orgánicas, este caso también es raro. Si, el compuesto no da una señal EPR. Todavía puede ser posible observar el estado de triplete de manera transitoria después de la fotoexcitación a un estado singlete excitado y el cruce entre sistemas al estado triplete.
Se observa acoplamiento de intercambio débil en biradicales con SOM bien localizados que se separan en escalas de longitud entre y. En tales casos, el acoplamiento de intercambio disminuye exponencialmente con la distancia entre los dos electrones o con el número de enlaces conjugados que separan los dos centros de densidad de espín. Si los dos centros no están unidos por una cadena continua de enlaces conjugados, el acoplamiento de intercambio rara vez se resuelve a distancias mayores que. En cualquier caso, a distancias tan largas el acoplamiento de intercambio es mucho menor que el acoplamiento dipolo-dipolo entre los dos electrones desapareados si el sistema no está conjugado. Para acoplamiento de intercambio débil, el sistema se describe más convenientemente en una representación desacoplada con dos giros y.
El acoplamiento de intercambio también es significativo durante los encuentros difusionales de dos moléculas paramagnéticas en solución líquida. Tal intercambio dinámico de espín de Heisenberg se puede representar como un intercambio físico de electrones desapareados entre las moléculas colisionantes. Esto provoca un cambio repentino del giro hamiltoniano, lo que lleva a la relajación del giro. Un ejemplo típico es el ensanchamiento de líneas en espectros EPR de radicales por oxígeno, que tiene un estado fundamental de triplete paramagnético. Si chocan radicales del mismo tipo, también se observa ensanchamiento de líneas, pero los efectos sobre los espectros pueden ser más sutiles, ya que los hamiltonianos de espín de los radicales colisionantes son los mismos. En este caso, el intercambio de electrones desapareados entre los radicales cambia solo el estado de espín, pero no el giro hamiltoniano.
14.5.2. Intercambio Hamiltoniano
La contribución hamiltoniana de giro por acoplamiento de intercambio débil es
Este hamiltoniano es análogo al hamiltoniano de acoplamiento en espectroscopía de RMN. Si los dos espines tienen valores diferentes y el campo es suficientemente alto, el intercambio Hamiltoniano se puede truncar de la misma manera que el Hamiltoniano de acoplamiento en RMN heteronuclear:
14.5.3. Manifestación espectral de la interacción de intercambio
En ausencia de acoplamiento hiperfino, la situación es la misma que para el acoplamiento en espectroscopía de RMN. El acoplamiento de intercambio entre espines similares (misma frecuencia de Zeeman de electrones) no influye en los espectros. Para los radicales en solución líquida, el acoplamiento hiperfino suele ser observable. En este caso, el acoplamiento de intercambio sí influye en los espectros incluso para espines similares, como se ilustra en la Figura para dos espines de electrones acoplados de intercambio y con cada uno de ellos acoplado exclusivamente a un solo espín nuclear y, respectivamente) con el mismo acoplamiento hiperfino. Si el acoplamiento de intercambio es mucho más pequeño que el acoplamiento isotrópico hiperfino, cada una de las líneas individuales del triplete hiperfino se divide aún más en tres líneas. Si la división es muy pequeña, puede notarse solo como un ensanchamiento de línea. En un acoplamiento de intercambio muy grande, los espines de electrones se distribuyen uniformemente sobre los dos restos acoplados a intercambio. De ahí que cada uno de ellos tenga el mismo acoplamiento hiperfino a ambos núcleos. Este acoplamiento es la mitad del acoplamiento hiperfino original, ya que, en promedio, el espín electrónico tiene solo la mitad de la densidad de espín en los orbitales de un núcleo dado en comparación con el caso sin acoplamiento de intercambio. Para los acoplamientos de intercambio intermedio, surgen patrones de división complejos que son característicos de la relación entre el acoplamiento de intercambio y el acoplamiento hiperfino.

Figura 5.1: Influencia del acoplamiento de intercambio sobre espectros EPR con acoplamiento hiperfino en solución líquida (simulación). Se muestran espectros para dos espines de electrones y con el mismo valor isotrópico y el mismo acoplamiento hiperfino isotrópico a un espín nuclear o, respectivamente. En ausencia de acoplamiento de intercambio, se observa un triplete con relación de amplitud. Para acoplamientos de intercambio pequeños, cada línea se divide en un triplete. En los acoplamientos de intercambio intermedio, resultan patrones complicados con muchas líneas. Para un acoplamiento de intercambio muy fuerte, cada espín de electrones se acopla a ambos núcleos de nitrógeno con la mitad del acoplamiento de intercambio isotrópico. Se observa un quintillizo con relación de amplitud.
15. Interacción dipolo-dipolo
15.5.4. Imagen física
La interacción dipolo-dipolo magnético entre dos espines de electrones localizados con momentos magnéticos y toma la misma forma que la interacción clásica entre dos dipolos de punto magnético. La energía de interacción
generalmente depende de los dos ángulos y que los dipolos puntuales incluyen con el vector entre ellos y del ángulo diedro (Figura 5.2). La interacción dipolo-dipolo escala con el cubo inverso de la distancia entre los dos dipolos puntuales.
En general, los dos espines de electrones están distribuidos espacialmente en sus respectivos SOM. La aproximación punto-dipolo sigue siendo una buena aproximación si la distancia es mucho mayor que la distribución espacial de cada espín electrónico. Una mayor simplificación es posible si la anisotropía es mucho menor que el valor isotrópico. En ese caso, los dos giros se alinean paralelos al campo magnético y por lo tanto también paralelos entre sí, de manera que y. Eq. (5.4) luego simplifica a
que es la forma conocida a partir de la espectroscopía de RMN.
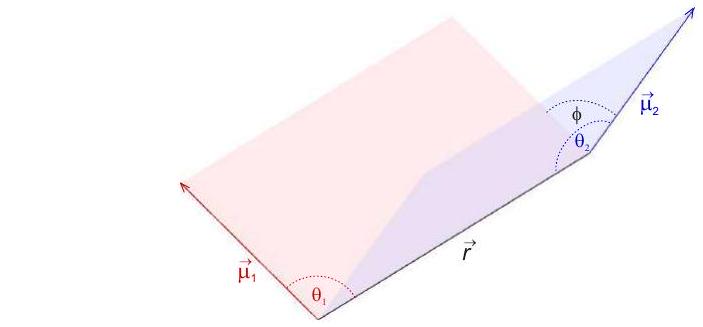
Figura 5.2: Geometría de dos dipolos magnéticos puntuales en orientación general. Los ángulos y se incluyen entre los respectivos vectores de momento magnético o y el vector de distancia entre los dipolos puntuales. Ángulo es el ángulo diedro.
15.5.5. Dipolo-dipolo Hamiltoniano
Para dos espines electrónicos que no están necesariamente alineados paralelos al campo magnético externo, el término de acoplamiento dipolo-dipolo del espín Hamiltoniano asume la forma
Si los electrones se distribuyen en el espacio, el hamiltoniano tiene que ser promediado (integrado) sobre las dos distribuciones espaciales, ya que el movimiento de electrones procede en una escala de tiempo mucho más rápida que un experimento EPR.
Si los dos electrones desapareados están bien localizados en la escala de longitud de sus distancias y sus espines están alineados paralelos al campo magnético externo, el dipolo-dipolo hamiltoniano toma la forma
con los términos del alfabeto dipolar
Por lo general, la espectroscopia EPR se realiza en campos donde la interacción Zeeman de electrones es mucho mayor que el acoplamiento dipolo-dipolo, que tiene una magnitud de aproximadamente a una distancia de y de a una distancia de. En esta situación, los términos, y son no seculares y pueden ser abandonados. El término es pseudo-secular y solo se puede eliminar si

Figura 5.3: Explicación del acoplamiento dipolo-dipolo entre dos espines en una imagen de campo local. En el giro del observador (azul) se induce un campo magnético local por el momento magnético del espín del compañero de acoplamiento (rojo). En la aproximación secular sólo es relevante el componente de este campo, que es paralelo o antiparalelo al campo magnético externo. La magnitud de este componente depende del ángulo entre el campo magnético externo y el vector spin-spin. Para los estados (izquierda) y (derecha) del giro del compañero, el campo local en el giro del observador tiene la misma magnitud, pero dirección opuesta. En la aproximación a altas temperaturas, ambos estados están igualmente poblados. El desplazamiento de la frecuencia de resonancia del giro del observador conduce así a una división de la transición del giro del observador, que es el doble del producto del campo local con la relación giromagnética del giro del observador.
la diferencia entre las frecuencias de Zeeman de electrones es mucho mayor que el acoplamiento dipolo-dipolo 1. En experimentos de doble resonancia electrónica electrónica (ELDOR), la diferencia de las frecuencias Larmor de los dos espines acoplados se puede seleccionar a través de la diferencia de las dos frecuencias de microondas. Por lo tanto, es posible excitar pares de espines para los que solo se necesita considerar la parte secular del giro hamiltoniano,
con
El acoplamiento dipolo-dipolo tiene entonces una simple dependencia del ángulo entre el campo magnético externo y el vector espín-espín y el acoplamiento puede interpretarse como la interacción del espín con el componente del campo magnético local que es inducido por el momento dipolo magnético del acoplamiento socio (Figura 5.3). Dado que el promedio del segundo polinomio de Legendre en todos los ángulos desaparece, la interacción dipolo-dipolo desaparece bajo un rápido movimiento isotrópico. Por lo tanto, las mediciones de esta interacción se realizan en estado sólido.
El tensor dipolo-dipolo en la aproximación secular tiene los valores propios. El acoplamiento dipolo-dipolo en cualquier orientación viene dado por
15.5.6. Manifestación espectral de la interacción dipolo-dipolo
El esquema de nivel de energía y un espectro esquemático para un par de espín con ángulo fijo se muestran en la Figura y b, respectivamente. Los acoplamientos dipolo-dipolo dividen la transición de cualquiera de los dos espines acoplados por. Si la muestra es macroscópicamente isotrópica, por ejemplo un polvo microcristalino o una solución congelada vítrea, todos los ángulos ocurren con probabilidad. Cada línea del doblete dipolar
El acoplamiento hiperfino de los espines electrónicos puede modificar esta condición.
Figura 5.4: Esquema de nivel de energía (a) y espectro esquemático (b) para un par de espín acoplado dipolo-dipolo en orientación fija con respecto al campo magnético. Las frecuencias de Zeeman de electrones de los dos espines son y, respectivamente. Se supone un acoplamiento débil. La división dipolar es la misma para ambos giros. Dependiendo del ancho de línea homogéneo, la división puede resolverse o no. Si y se distribuyen, por ejemplo por anisotropía, la resolución se pierde incluso para.
luego se ensancha a un patrón de polvo como se ilustra en la Figura 3.3. El patrón de polvo para el estado del giro asociado es una imagen especular de la del estado, ya que los desplazamientos de frecuencia por el campo magnético local tienen signo opuesto para los dos estados. La superposición de los dos patrones axiales de polvo se denomina patrón Pake (Figura 5.5). El centro del patrón Pake corresponde al ángulo mágico. El acoplamiento dipolo-dipolo se desvanece en este ángulo.

Figura 5.5: Patrón de Pake observado para un par de espín acoplado dipolo-dipolo. (a) La división del doblete dipolar varía según el ángulo entre el vector spin-spin y el campo magnético estático. Las orientaciones tienen probabilidad. (b) La suma de todos los dobletes para una distribución uniforme de las direcciones del vector espín-espín es el patrón Pake. Los “cuernos” están divididos por y los “hombros” están divididos por. El centro del patrón corresponde al ángulo mágico.
El patrón de Pake se observa muy raramente en un espectro EPR, ya que generalmente otras interacciones anisotrópicas son mayores que la interacción dipolo-dipolo entre espines de electrones. Si la condición de acoplamiento débil se cumple para la gran mayoría de todas las orientaciones, la forma de línea EPR se aproxima bien por una convolución del patrón Pake con la forma de línea en ausencia de interacción dipolo-dipolo. Si se conoce esta última forma de línea, por ejemplo a partir de la medición de muestras análogas que transportan solo uno de los dos espines de electrones, el patrón de Pake se puede extraer por deconvolución y la distancia entre los dos espines electrónicos se puede inferir de la división invirtiendo la Ec. (5.15).
15.1. Interacción de campo cero
15.1.1. Imagen física
Si varios giros no emparejados están muy fuertemente acoplados de intercambio, entonces son mejor descritos por un giro grupal. El concepto es más fácil de entender para el caso de dos espines electrónicos que ya hemos discutido en la Sección 5.1.1. En este caso, el estado singlete con espín grupal es diamagnético y por lo tanto no observable por EPR. Los tres subniveles del estado de triplete observable con espín grupal corresponden a números cuánticos magnéticos, y a campo alto. Estos niveles se dividen por la interacción de Zeeman de electrones. Las transiciones y son transiciones de espín electrónico permitidas, mientras que la transición es una transición doble cuántica prohibida.
En el campo magnético cero, la interacción de Zeeman de electrones desaparece, sin embargo, los tres subniveles de tripletes no son degenerados, exhiben división de campo cero. Esto se debe a que los electrones desapareados también están acoplados dipolo-dipolo. La integración de la Ec. (5.6) sobre la distribución espacial de los dos espines de electrones en sus respectivos SOM proporciona un tensor de interacción de campo cero que puede ser fundido en una forma donde describe el acoplamiento del espín del grupo consigo mismo [Rie07]. En el campo cero, los subniveles de triplete no son descritos por el número cuántico magnético, que es un buen número cuántico solo si la interacción de Zeeman de electrones es mucho mayor que la interacción de campo cero. Más bien, los subniveles de triplete en el campo cero están relacionados con las direcciones de los ejes principales del tensor de interacción de campo cero y por lo tanto se etiquetan, y, mientras que los subniveles en la aproximación de campo alto están etiquetados, y.
Este concepto puede extenderse a un número arbitrario de espines de electrones fuertemente acoplados. Los casos con hasta 5 electrones desapareados fuertemente acoplados ocurren para iones de metales de transición (d shell) y los casos con hasta 7 electrones desapareados fuertemente acoplados ocurren para iones de tierras raras (f shell). Según la regla de Hund, en ausencia de un campo ligando el estado con mayor espín grupal es el estado fundamental. Los iones Kramers con un número impar de electrones desapareados tienen un giro de grupo medio entero. Se comportan de manera diferente a los iones que no son Kramers con un número par de electrones y un giro de grupo entero. Esta clasificación se relaciona con el teorema de Kramers, que establece que para un sistema simétrico de inversión de tiempo con giro total de medio entero, todos los autoestados ocurren como pares (pares Kramers) que son degenerados en campo magnético cero. Como consecuencia, para los iones Kramers el estado fundamental en campo cero se dividirá cuando se aplique un campo magnético. Para cualquier frecuencia de microondas existe un campo magnético donde la transición dentro del doblete Kramers de tierra es observable en un espectro EPR. Lo mismo no se aplica para el giro de grupo entero, donde el estado fundamental no puede ser degenerado en campo cero. Si la interacción de campo cero es mayor que la frecuencia de microondas máxima disponible, los iones no KRAMers pueden ser inobservables por espectroscopia EPR aunque existen en un estado paramagnético de alto espín. Los ejemplos típicos de tales iones silenciosos que no son KRAMers de EPR son de alto espín y alto espín. En casos raros, los iones no KRAMers son EPR observables, ya que el estado fundamental puede degenerar en campo magnético cero si el campo ligando presenta simetría axial. Tenga en cuenta también que los iones “silenciosos EPR” que no son Kramers pueden llegar a ser observables a una frecuencia de microondas y campo magnético suficientemente altos.
Para los iones de metales de transición y tierras raras, la interacción de campo cero no se debe únicamente a la interacción dipolo-dipolo entre los espines de electrones. El acoplamiento espín-órbita también contribuye, en muchos casos incluso más fuerte que la interacción dipolo-dipolo. La predicción cuántico-química de la interacción de campo cero es un campo activo de investigación. Se pueden obtener predicciones bastante razonables para los iones de metales de transición, mientras que solo las estimaciones de orden de magnitud suelen ser posibles para los iones de tierras raras.
15.1.2. Interacción de campo cero Hamiltoniano
La interacción de campo cero Hamiltoniano a menudo se da como
donde denota la transposición del operador de vector de giro. En el sistema de ejes principales del tensor de división de campo cero (ZFS), el hamiltoniano simplifica
dónde y. La reducción a dos parámetros es posible, ya que es un tensor sin rastro. En otras palabras, la interacción de campo cero es puramente anisotrópica. La notación presume que es el valor principal con el mayor valor absoluto puede ser negativo). Junto con la ausencia de un componente isotrópico, esto significa que, que es siempre el valor intermedio, está más cerca que o exactamente en el medio entre estos dos valores. En consecuencia,. En simetría axial. La simetría axial se aplica si el sistema tiene un eje de simetría con. En simetría cúbica, ambos y son cero. Para el giro grupal, el término principal del es entonces una contribución hexadecapolar que escala con la cuarta potencia de los operadores de centrifugado.
En la aproximación de campo alto la contribución de ZFS al hamiltoniano es un término. En otras palabras, a primer orden en la teoría de la perturbación la contribución de la ZFS a la energía de un nivel de espín con escalas numéricas cuánticas magnéticas con. Para una transición permitida, esta contribución es. Esta contribución se desvanece para la transición central de los iones Kramers. De manera más general, debido al escalado de las energías de nivel con a primer orden, la contribución de ZFS a las frecuencias de transición desaparece para todas las transiciones.
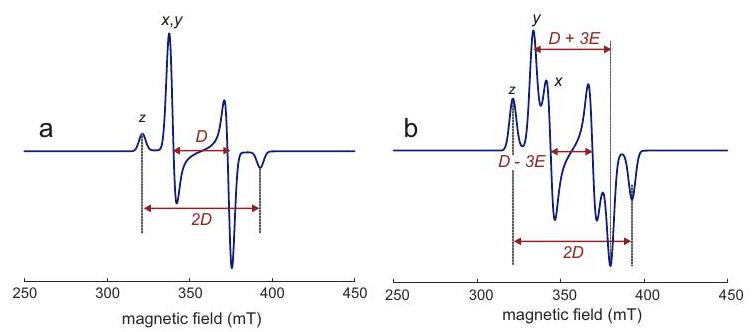
Figura 5.6: Espectros CW EPR esquemáticos para estados tripletes en campo alto. Las simulaciones se realizaron a una frecuencia de banda X de. a) Simetría axial. El espectro es la derivada de un patrón de Pake. b) Simetría ortorrómbica.
15.1.3. Manifestación espectral de división de campo cero
Los espectros son más fáciles de entender en la aproximación de campo alto. Muy a menudo, las desviaciones de esta aproximación son significativas para el ZFS (ver Fig. 2.2), y tales desviaciones se discuten más adelante. El otro caso limitante, donde la ZFS es mucho mayor que la interacción de Zeeman de electrones (Fe (III) y la mayoría de los iones de tierras raras), se discute en la Sección 5.3.4.
Para estados tripletes con simetría axial del tensor ZFS, el espectro de absorción es un patrón Pake (ver Sección 5.2.3). Con EPR de onda continua se detecta la derivada del espectro de absorción, el cual tiene la apariencia mostrada en la Fig. 5.6 (a). Una desviación de la simetría axial conduce a una división de los “cuernos” del patrón Pake por, mientras que los “hombros” del patrón no se ven afectados (Fig. 5.6 (b)). Los estados tripletes de las moléculas orgánicas a menudo se observan después de la excitación óptica de un estado singlete y el cruce entre sistemas. Dicho cruce entre sistemas generalmente conduce a diferentes poblaciones de los subniveles de triplete de campo cero, y. En esta situación el sistema de espín no está en equilibrio térmico, sino polarizado por espín. Dicha polarización de espín afecta la intensidad relativa de las singularidades de la forma de línea en los espectros e incluso el signo de la señal puede cambiar. Sin embargo, las singularidades aún se observan en los mismos campos de resonancia, es decir, los parámetros y aún se pueden leer de los espectros como se indica en la Fig.
Incluso si las poblaciones de los subniveles triples se han relajado al equilibrio térmico, el espectro aún puede diferir del espectro de aproximación de campo alto, como se ilustra en la Fig. para el triplete de naftaleno excitado (simulación realizada con un script de ejemplo del paquete de software EasySpin http://WWW. easyspin.org/). Porque a un campo de aproximadamente (frecuencia Zeeman de electrones de aproximadamente) se viola la aproximación de campo alto y ya no es un buen número cuántico. De ahí que la transición doble cuántica formalmente prohibida se haga parcialmente permitida. A primer orden en la teoría de la perturbación, esta transición no es ampliada por la ZFS. Por lo tanto, es muy estrecho en comparación con las transiciones permitidas y aparece con mayor amplitud.
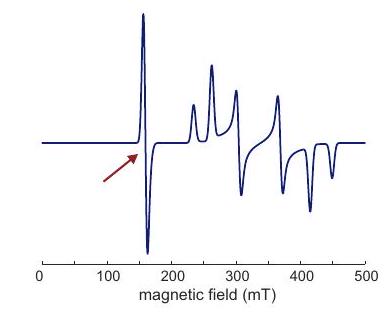
Figura 5.7: Espectro CW EPR del triplete de naftaleno excitado en equilibrio térmico (simulación a una frecuencia de banda de). La flecha roja marca la señal de medio campo, que corresponde a la transición doble cuántica formalmente prohibida.
Para los iones Kramers, los espectros suelen estar dominados por la transición central, que no está ensanchada por ZFS a primer orden. A segundo orden en la teoría de la perturbación, el ensanchamiento ZFS de esta línea escala inversamente con el campo magnético. Por lo tanto, mientras que los sistemas con anisotropía exhiben ensanchamiento proporcional al campo magnético, las transiciones centrales de los iones Kramers muestran estrechamiento con. Estos últimos sistemas pueden ser detectados con una sensibilidad extremadamente alta en campos altos si no presentan anisotropía significativa. Esto se aplica a sistemas con conchas medio llenas (e.g.). En el caso de Mn (II) (Figura 5.8) la transición central estrecha se divide en seis líneas por acoplamiento hiperfino al espín nuclear de (abundancia natural de espín nuclear). Debido al escalado de la ampliación anisotrópica de las transiciones ZFS, las transiciones satelitales se vuelven cuanto más amplias son las mayores para los niveles involucrados. En la aproximación a altas temperaturas, la intensidad integral en el espectro de absorción es la misma para todas las transiciones. De ahí que las transiciones más amplias hagan una menor contribución a la amplitud en el espectro de absorción y en su primera derivada que es adquirida por CW EPR.

Figura 5.8: Espectro CW EPR de un complejo Mn (II) (simulación a una frecuencia de banda W de). Las seis líneas estrechas intensas son el multiplete hiperfino de la transición central.
La situación puede complicarse aún más por y cepa, que es una distribución de los parámetros y debido a una distribución en el campo del ligando. Tal caso se demuestra en la Fig. para Gd (III) a una frecuencia de microondas de donde la ampliación de segundo orden de la transición central sigue siendo bastante fuerte. En tal caso, las singularidades de la forma de línea se lavan y los parámetros ZFS no se pueden leer directamente de los espectros. En CW EPR, las transiciones satelitales pueden permanecer inobservadas ya que la derivada de la forma de línea de absorción es muy pequeña excepto por la transición central.
15.1.4. Giro efectivo en dobletes de Kramers
Para algunos sistemas, como el Fe (III), ZFS es mucho más grande que la interacción del electrón Zeeman en cualquier campo magnético experimentalmente alcanzable. En este caso, la interacción de campo cero determina la dirección de cuantificación y la interacción de Zeeman de electrones puede tratarse como una perturbación [Cas+60]. El tratamiento es más sencillo para la simetría axial, donde el eje de cuantificación es el eje del tensor ZFS. Las energías en ausencia del campo magnético son
que para Fe (III) de alto giro da tres dobletes Kramers degenerados correspondientes a, y. Si el campo magnético se aplica a lo largo del eje del tensor ZFS, es un buen número cuántico y simplemente hay un término de energía adicional con ser el valor para la concha medio llena, que puede aproximarse como. Además, en esta situación sólo se permite la transición. El término Zeeman lleva a una división del doblete de los Kramers que es proporcional a y
Figura 5.9: Espectro EPR ecodetectado (espectro de absorción) de un complejo Gd (III) con GHz, una distribución gaussiana de con desviación estándar de y una distribución correlacionada de (simulación a una frecuencia de banda Q por cortesía del Dr. Maxim Yulikov). (a) Espectro total (negro) y contribuciones de las transiciones individuales (ver leyenda). La señal de la transición central (azul) domina. b) Contribuciones de las transiciones satelitales escaladas verticalmente para mayor claridad.
corresponde a. Este doblete de Kramers puede describirse así como un giro efectivo con.
Si el campo magnético es perpendicular al eje del tensor, los dobletes y Kramers no se dividen, ya que el operador y no conecta estos niveles. El operador tiene un elemento fuera de la diagonal que conecta los niveles que es. Dado que los niveles son degenerados en ausencia de la interacción del electrón Zeeman, se cuantifican a lo largo del campo magnético y nuevamente es un buen número cuántico de este doblete de Kramers. Las energías son, de manera que la frecuencia de transición vuelve a ser proporcional a, pero ahora con un valor efectivo. Las orientaciones intermedias se pueden describir asumiendo un tensor efectivo con simetría axial y. Esta situación se encuentra con una buena aproximación para Fe (III) de alto espín en hemoglobinas.
Para el caso no axial, el campo magnético dividirá los tres dobletes Kramers. A primer orden en la teoría de la perturbación la división es proporcional a, lo que significa que cada doblete de Kramers puede describirse mediante un giro efectivo con un tensor efectivo. Otro caso sencillo se encuentra por rombocidad extrema,. Al reordenar los ejes principales (intercambiando con cualquiera o) uno puede deshacerse del término en la ecuación (5.18), de manera que el ZFS hamiltoniano reduce a con. El par de niveles correspondiente a la nueva dirección del tensor tiene energía cero a campo magnético cero y se puede demostrar que tiene un valor efectivo isotrópico. De hecho, las señales cercanas se observan muy a menudo para el Fe (III) de alto giro.


15.2. Imagen física
15.2.1. El sistema de centrifugado
Los fenómenos básicos se pueden entender bien en el sistema de espín electrón-nuclear más simple posible que consiste en un espín de un solo electrón con valor isotrópico que es hiperfino acoplado a un espín nuclear con una magnitud del acoplamiento hiperfino que es mucho menor que la interacción de Zeeman del electrón. En esta situación la aproximación de campo alto es válida para el espín electrónico, de manera que el hamiltoniano hiperfino puede ser truncado a la forma dada por la Ec. (4.9). Debido a la ocurrencia de un operador en este hamiltoniano, no podemos simplemente transformar al hamiltoniano en el marco giratorio para el giro nuclear. Sin embargo, no es necesario, ya que consideraremos solo la irradiación de microondas. Para el espín electrónico, transformamos al marco giratorio donde este giro tiene un desplazamiento de resonancia. De ahí que el total hamiltoniano tome la forma
en el marco giratorio para el espín electrónico y el marco de laboratorio para el espín nuclear. Tal hamiltoniano es una buena aproximación, por ejemplo, para protones en radicales orgánicos.
El hamiltoniano se desvía del hamiltoniano que se aplicaría si también se cumpliera la aproximación de campo alto para el giro nuclear. La diferencia es el término de acoplamiento hiperfino pseudo-secular. Como puede verse en la Ec. (4.10), este término desaparece si la interacción hiperfina es puramente isotrópica, es decir, para voltear suficientemente rápido en solución líquida, y a lo largo de los ejes principales del tensor hiperfino. De lo contrario, el término sólo se puede descuidar si, correspondiente a la aproximación de campo alto del espín nuclear. Dentro del rango aproximado la interacción pseudo-secular puede afectar las frecuencias de transición y realiza transiciones formalmente prohibidas con parcialmente permitidas, ya que ya no es un buen número cuántico.
15.2.2. Campos locales en el giro nuclear
La ocurrencia de transiciones prohibidas se puede entender en una imagen vectorial de magnetización semi-clásica considerando campos locales en el espín nuclear para los dos estados posibles y
El producto del tiempo de correlación rotacional y la anisotropía hiperfina debe ser mucho menor que la unidad

Figura 6.1: Campos locales (multiplicados por la relación giromagnética del espín nuclear) en el espín nuclear en los dos estados y de un espín electrónico. Los ejes de cuantificación están a lo largo de los campos efectivos y y, por lo tanto, no son paralelos.
del espín electrónico. Estos campos locales se obtienen de los parámetros, y de los términos del operador Hamilton que actúan sobre el giro nuclear. Cuando se dividen por la relación giromagnética del espín nuclear, estos términos tienen la dimensión de un campo magnético local. El campo local correspondiente a la interacción nuclear de Zeeman es igual al campo magnético estático y es el mismo para ambos estados de espín electrónico, ya que el valor de expectativa de no depende del estado de espín electrónico. Se alinea con la dirección del marco del laboratorio (flecha azul en la Figura 6.1). Ambos campos hiperfinos surgen de términos hamiltonianos que contienen un factor, que tiene el valor de expectativa para el estado y para el estado. El término se alinea con el eje y se dirige hacia en el estado y hacia en el estado, asumiendo (flechas violetas). El término se alinea con el eje y se dirige hacia en el estado y hacia en el estado, asumiendo (flechas verdes).
Los campos efectivos en el espín nuclear en los dos estados de espines electrónicos son sumas vectoriales de los tres campos locales. Debido al componente a lo largo, se inclinan desde la dirección por ángulo en el estado y por ángulo en el estado. La longitud de los vectores suma son las frecuencias de transición nuclear en estos dos estados y están dadas por
Para, la división hiperfina viene dada por
y la frecuencia suma viene dada por
Para, el doblete de frecuencia nuclear está centrado en la Fig.). La frecuencia suma es siempre mayor que el doble de la frecuencia nuclear Zeeman. Ninguna de las frecuencias nucleares puede llegar a ser cero, el valor mínimo posible se alcanza en uno de los estados de espín electrónico para el emparejamiento de la interacción nuclear Zeeman e hiperfina en. Para la frecuencia nuclear, el doblete se divide por y se centra en la Fig. Los ángulos de inclinación y (Figura 6.1) se pueden inferir de las relaciones trigonométricas y están dados por
Consideremos ahora una situación en la que el espín electrónico está en su estado. La magnetización nuclear de todos los radicales en este estado en equilibrio térmico está alineada con. La excitación de microondas provoca transiciones al estado. En este estado, el campo local en el giro nuclear se dirige a lo largo. Por lo tanto, el vector de magnetización nuclear de los radicales bajo consideración se inclina en ángulo (Figura 6.1) con respecto al campo local actual. Comenzará a preceder alrededor de este vector de campo local. Esto corresponde a la excitación del espín nuclear al voltear el espín electrónico, que es una transición formalmente prohibida. Obviamente, tal excitación ocurrirá sólo si el ángulo difiere de y de. El caso de corresponde a la ausencia de acoplamiento hiperfino pseudo-secular y también se alcanza en el límite. La situación se alcanza en el límite de acoplamiento hiperfino secular muy fuerte,. Se observan transiciones prohibidas para el acoplamiento hiperfino intermedio. Se espera una excitación máxima de espines nucleares cuando los dos ejes de cuantificación son ortogonales entre sí,.
Figura 6.2: Sistema de espín electrón-nuclear en presencia de acoplamiento hiperfino pseudo-secular. a) Esquema de niveles. En EPR se permiten las transiciones (rojo), en RMN se permiten las transiciones (azul), y las transiciones cero y dobles cuánticas con están formalmente prohibidas. (b) Espectro de varilla EPR. Las transiciones permitidas tienen probabilidad de transición y probabilidad de transiciones prohibidas. El espectro se muestra para. Porque, las transiciones prohibidas se encuentran dentro del doblete de transición permitido. (c) Espectro de RMN para. (d) Espectro de RMN para.
15.3. Formalismo operador de producto con interacciones pseudo-seculares
15.3.1. Transformación de a la propia base
La excitación y detección en experimentos de EPR son descritas por los operadores y en el marco giratorio. Estos operadores actúan únicamente sobre las transiciones de espín electrónico y así formalizan las reglas de selección espectroscópica. Si el giro hamiltoniano contiene términos fuera de diagonal, como el término pseudo-secular en la ecuación (6.1), la base propia se desvía de la base del marco giratorio de espín electrónico/marco de laboratorio de espín nuclear en el que está escrito el hamiltoniano y en el que los operadores de excitación y detección son lineales combinaciones de y. Para entender qué transiciones son impulsadas y detectadas con qué momento de transición, necesitamos transformarnos a la base propia (la transformación de es análoga). Esto se puede hacer por formalismo operador de producto y se puede entender en la imagen de campo local.
El hamiltoniano en la base propia no tiene elementos fuera de la diagonal, lo que significa que todos los ejes de cuantificación están a lo largo. Así, podemos inferir directamente de la Fig. que, en el estado, necesitamos una rotación en sentido contrario a las agujas del reloj (matemáticamente positiva) por ángulo de inclinación alrededor del eje, que está apuntando hacia el plano del papel. En el estado, necesitamos una rotación en sentido horario (matemáticamente negativa) por ángulo de inclinación alrededor del eje. Los estados de espín electrónico pueden ser seleccionados por los operadores de proyección y, respectivamente. De ahí, tenemos que aplicar rotaciones y. Estas dos rotaciones viajan, ya que los subespacios y son distintos cuando es un buen número cuántico. Para la rotación en la base propia, podemos escribir una matriz unitaria
dónde y. Obsérvese que la definición de ángulo corresponde a la dada gráficamente en la Fig. 6.1.2 Las dos nuevas rotaciones alrededor y también conmutan. Además, se desplaza con y), de manera que la transformación de a la base propia se reduce a
El momento de transición para las transiciones permitidas que son impulsadas por se multiplica por un factor, es decir, se vuelve más pequeño cuando. Para interpretar el segundo término, se reescribe mejor en términos de operadores de escalera y. ENCONTRAMOS
En otras palabras, este término impulsa las transiciones electrón-nuclear cero y doble cuánticas prohibidas (Fig. 6.2 (a)) con una transición proporcional a.
En un experimento CW EPR, cada transición debe ser excitada y detectada. En otras palabras, la amplitud es proporcional al cuadrado del momento de transición, que es la probabilidad de transición. Las transiciones permitidas tienen así una intensidad proporcional y las transiciones prohibidas una probabilidad de transición proporcional a (Fig. 6.2 (b)).
15.3.2. Cálculo general del operador de producto para un Hamiltoniano no diagonal
En un cálculo de operador de producto, los términos del hamiltoniano se pueden aplicar uno tras otro si y solo si viajan por pares. Este no es el caso del hamiltoniano en la Ec. (6.1). Sin embargo, la aplicación de diagonaliza el hamiltoniano:
Esto proporciona una receta simple para los cálculos del operador del producto en presencia del acoplamiento hiperfino pseudosecular. La evolución libre y los pulsos selectivos de transición se calculan en el
Hemos utilizado y. eigenbasis, usando el hamiltoniano en el lado derecho de la relación (6.9). Para la aplicación de pulsos no selectivos, el operador de densidad necesita ser transformado a la base de marco giratorio de espín electrónico/marco de laboratorio de espín nuclear aplicando. En el formalismo operador de producto esto corresponde a una transformación de operador de producto. Después de la aplicación de pulsos no selectivos, el operador de densidad necesita ser retrotransformado a la propia base. La detección también debe realizarse en la base del marco de rotación de espín electrónico/marco de laboratorio de espín nuclear.
Este concepto se puede extender a cualquier hamiltoniano no diagonal siempre y cuando se pueda encontrar una transformación unitaria que transforme el hamiltoniano a su base propia y se pueda expresar mediante un solo término de operador de producto o una suma de términos de operador de producto de desplazamiento por pares.
15.4. Generación y detección de coherencia nuclear por excitación de espín electrónico
15.4.1. Generador de coherencia nuclear
Hemos visto que un solo pulso de microondas puede excitar la coherencia en transiciones prohibidas electrón-nuclear cero y doble cuántica. En principio, esto proporciona acceso a las frecuencias nucleares y, que son diferencias de frecuencias de transiciones de espín electrónico permitidas y prohibidas, como se puede inferir de la Fig. 6.2 (a, b). En efecto, la decaimiento de un eco de Hahn de espín electrónico como una función de se modula con frecuencias y así como con y). Dicha modulación surge de transiciones prohibidas durante el pulso de reenfoque, que redistribuyen la coherencia entre las cuatro transiciones. Los ecos de transferencia de coherencia son modulados por la diferencia de las frecuencias de resonancia antes y después de la transferencia por el pulso, en las que se cancela el desplazamiento de resonancia, mientras que las contribuciones de espín nuclear no cancelan. Este experimento ESEEM de dos pulsos no suele aplicarse para medir acoplamientos hiperfinos, ya que la aparición de las frecuencias de combinación y complica los espectros y el ancho de línea se determina por la relajación transversal de espín electrónico, que es mucho más rápida la relajación transversal del espín nuclear.
Se puede obtener una mejor resolución y espectros más simples mediante la observación indirecta de la evolución de la coherencia nuclear. Dicha coherencia se puede generar aplicando primero un pulso a los espines electrónicos, lo que generará coherencia de espín electrónico en transiciones permitidas con amplitud proporcional a y sobre transiciones prohibidas con amplitud proporcional al pecado. Después de un retraso se aplica un segundo pulso. Obsérvese que el bloque es parte de los experimentos EXSY y NOESY en RMN. El segundo pulso generará un componente de magnetización de espín electrónico a lo largo de la mitad de la coherencia de espín electrónico existente, es decir, “apagará” la mitad de la coherencia de espín electrónico y la convertirá en polarización. Sin embargo, para la coherencia en las transiciones prohibidas, existe la posibilidad de que el espín nuclear no sea volteado, es decir, que sobreviva la superposición coherente de los estados de espín nuclear. Para la coherencia de espín electrónico en las transiciones permitidas, existe la posibilidad de que la “desconexión” de la coherencia electrónica conduzca a un “encendido” de las coherencias nucleares. De ahí que en ambas vías exista una probabilidad proporcional a que se genere coherencia nuclear. Se requiere el retraso, ya que en los diferentes componentes de coherencia nuclear tienen fase opuesta y cancelan.
La coherencia nuclear generada por el bloque puede ser calculada por formalismo operador producto como se describe en la Sección 6.2.2. ENCONTRAMOS
Esta expresión se puede interpretar de la siguiente manera. La coherencia nuclear se crea con una fase como si hubiera comenzado a evolucionar como en el tiempo (últimos factores coseno en el lado derecho de cada línea). Se modula como una función del desplazamiento de resonancia de espín electrónico y cero exactamente en la resonancia (primer factor en cada línea). La integral sobre una línea EPR inhomogéneamente ensanchada y simétrica también es cero, ya que. Sin embargo, esto puede ser compensado posteriormente aplicando otro pulso. La amplitud de la coherencia nuclear generalmente escala con, ya que se requiere una transferencia permitida y otra prohibida para excitarla y (segundo factor). El tercer factor en el lado derecho de las líneas 1 y 2 dice que la amplitud de la coherencia con frecuencia se modula en función de con frecuencia. Asimismo, la amplitud de la coherencia con frecuencia se modula en función de con frecuencia (líneas 3 y 4). A ciertos valores de no coherencia se crea en la transición con frecuencia, en otros momentos se genera máxima coherencia. Tal comportamiento se llama comportamiento de punto ciego. Para detectar todas las frecuencias nucleares, un experimento basado en el generador de coherencia nuclear tiene que repetirse para diferentes valores de. Por qué y cómo se realiza la espectroscopia CW EPR Ventajas de sensibilidad de la espectroscopia CW EPR El experimento CW EPR
Consideraciones sobre la preparación de muestras Descripción teórica de CW EPR Spin packet lineshape Saturation
16. 7 - Espectroscopia CW EPR
16.1. Por qué y cómo se realiza la espectroscopia CW EPR
16.1.1. Ventajas de sensibilidad de la espectroscopia CW EPR
En espectroscopía de RMN, las técnicas de CW han sido casi completamente desplazadas por las técnicas de transformada de Fourier (FT), excepto por algunas aplicaciones de nicho. Las técnicas FT tienen una ventaja de sensibilidad si el espectro contiene grandes secciones de línea base y todo el espectro puede ser excitado simultáneamente por los pulsos. Ninguna de las condiciones suele cumplirse en la espectroscopia EPR. Por dos razones, las técnicas de FT pierden sensibilidad en la espectroscopia EPR en comparación con el experimento CW. Primero, mientras que los espectros de RMN típicos encajan cómodamente en el ancho de banda de un circuito de resonancia de radiofrecuencia acoplado críticamente bien diseñado, los espectros EPR son mucho más amplios que el ancho de banda de un resonador de microondas con factor de alta calidad. Ampliar el ancho de banda de detección y reducir proporcionalmente el factor de calidad del resonador reduce la relación señal/ruido a menos que la línea de absorción sea infinitamente amplia. Un factor de calidad del orden de 10 '000, que se puede lograr con resonadores de cavidad, corresponde a un ancho de banda de aproximadamente a frecuencias de banda X alrededor. La alta sensibilidad intrínseca de detección en una banda tan estrecha solo puede usarse en un experimento de CW. En segundo lugar, incluso si el resonador está sobreacoplado a un factor de calidad mucho más bajo o se utilizan resonadores con intrínsecamente más bajos (la pérdida de sensibilidad puede compensarse parcialmente por un factor de llenado más alto de tales resonadores), la potencia residual de un pulso de microondas de alta potencia requiere aproximadamente para disminuir por debajo el nivel de una señal EPR. Este tiempo muerto suele ser una fracción significativa del tiempo de relajación transversal de los espines electrónicos, lo que conlleva la pérdida de señal por relajación. En contraste, en la espectroscopia de RMN el tiempo muerto suele ser despreciablemente corto en comparación con los tiempos de relajación. En muchos casos, el tiempo muerto en la espectroscopia de EPR pulsada supera incluso con fuerza. En esta situación FT EPR es imposible, incluso con el reenfoque de eco, mientras que los espectros CW EPR aún se pueden medir. Este caso suele aplicarse a complejos de metales de transición a temperatura ambiente y a muchos complejos de metales de tierras raras y complejos de Fe (III) de alto espín incluso hasta el punto de ebullición del helio líquido a presión normal (4.2 K). Por estas razones, cualquier muestra desconocida potencialmente paramagnética debe caracterizarse primero por espectroscopía CW EPR. Se requieren técnicas de EPR pulsadas si la resolución de la espectroscopia CW EPR proporciona información insuficiente para asignar una estructura. Esto se aplica principalmente a pequeños acoplamientos hiperfinos en radicales orgánicos y de núcleos de ligandos en complejos de metales de transición (ver Capítulo 8) y a la medición de distancias entre espines de electrones en el rango nanométrico (ver Capítulo 9). A temperaturas donde se pueden obtener señales de pulso EPR, la medición de los tiempos de relajación también es más fácil y precisa con técnicas de EPR pulsadas.
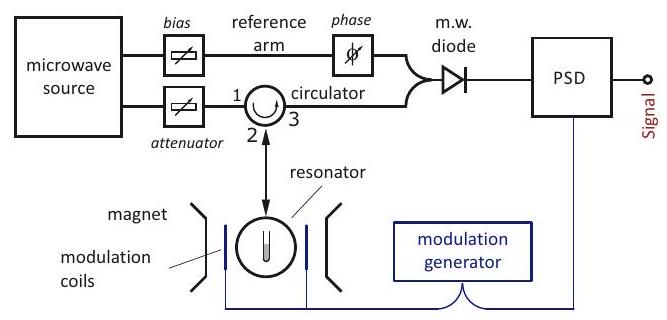
Figura 7.1: Esquema de un espectrómetro CW EPR. El microondas de una fuente de frecuencia fija se pasa a través de un atenuador para ajustar su potencia y luego a través de un circulador a la muestra. El microondas que regresa de la muestra pasa de una manera diferente a través del mismo circulador y se combina con microondas de referencia de potencia ajustable (polarización) y fase antes de que sea detectado por un diodo de microondas. La señal de salida de este diodo ingresa a un detector sensible a la fase (PSD) donde se desmodula con respecto a la frecuencia de modulación de campo (típicamente) y al mismo tiempo se amplifica. La señal de salida del PSD se digitaliza y se procesa posteriormente en una computadora. El espectro se obtiene barriendo el campo magnético estático a frecuencia constante de microondas.
16.1.2. El experimento CW EPR
Dado que el ancho de banda de un resonador de microondas optimizado es mucho menor que el ancho típico de los espectros EPR, no es práctico barrer la frecuencia en un campo magnético constante para obtener un espectro. En cambio, la frecuencia de microondas se mantiene constante y coincide con la frecuencia del resonador en todo momento. La condición de resonancia para los giros se establece barriendo el campo magnético. Otra dificultad surge del débil acoplamiento magnético de los giros al excitante campo electromagnético. Por lo tanto, solo se observa una fracción muy pequeña de la potencia de excitación. Este problema se resuelve de la siguiente manera. En primer lugar, se evita la transmisión directa de la potencia de excitación al detector mediante un circulador (Figura 7.1). La energía que ingresa al puerto 1 solo puede salir a la muestra a través del puerto 2. La potencia que viene de la muestra a través del puerto 2 solo puede dejar al diodo detector a través del puerto 3. Segundo, el resonador está acoplado críticamente. Esto significa que toda la potencia de microondas procedente de la fuente que incide en el resonador entra en el resonador y es convertida en calor por la impedancia (resistencia compleja) del resonador. Si la muestra está apagada resonante y por lo tanto no absorbe microondas, ninguna energía de microondas sale del resonador a través del puerto 3. Si ahora el campo magnético se ajusta a la condición de resonancia y la muestra absorbe resonantemente microondas, esto significa que la impedancia del resonador + muestra ha cambiado. El resonador ya no está acoplado críticamente y parte del microondas entrante se refleja. Este microondas sale del circulador a través del puerto 3 y es incidente en el diodo detector.
Esta potencia reflejada en la absorción de resonancia puede ser muy débil a una concentración de muestra baja. Por lo tanto, es importante detectarlo con sensibilidad. Un diodo de microondas solo es débilmente sensible a un cambio en la potencia incidente a baja potencia (Fig. 7.2, el voltaje de entrada es proporcional a la raíz cuadrada de la potencia). El diodo es más sensible a los cambios de amplitud cerca de su punto de funcionamiento, marcado en verde en la Fig. 7.2. Por lo tanto, el diodo debe estar polarizado a su punto de funcionamiento mediante la adición de potencia constante desde un brazo de referencia. La fase del brazo de referencia debe ajustarse para que el microondas procedente del resonador y el microondas procedente del brazo de referencia interfieran constructivamente.

Figura 7.2: Curva característica de un diodo de detección de microondas. A un voltaje de entrada pequeño, el diodo es bastante insensible a los cambios en el voltaje de entrada. En el punto de operación (verde), la dependencia de la corriente de salida de la tensión de entrada es lineal y tiene pendiente máxima. Esto corresponde a la corriente de salida. Si el voltaje de entrada es demasiado grande, el diodo se destruye (punto rojo).
Otro problema surge del hecho de que los diodos de microondas son detectores de banda ancha. Por un lado, esto es útil, ya que las muestras pueden desplazar significativamente la frecuencia del resonador. Por otro lado, los detectores de banda ancha también recogen ruido de una amplia banda de frecuencias. Esto disminuye la relación señal/ruido y debe contrarrestarse limitando el ancho de banda de detección al ancho de banda de la señal o incluso por debajo. Dicha limitación de ancho de banda se puede realizar más fácilmente mediante modulación de efectos y detección sensible a la fase. Al aplicar una modulación de campo magnético sinusoidal pequeña con frecuencia típica y amplitud típica de, el componente de señal en la salida del diodo detector se modula con la misma frecuencia, mientras que el ruido no está correlacionado con la modulación. La demodulación con una señal de referencia del generador de modulación de campo (Figura 7.1) por un detector sensible a la fase amplifica la señal y limita el ancho de banda a la frecuencia de modulación.
La modulación de efectos con detección sensible a fase mide la derivada de la línea de absorción, siempre y cuando la amplitud de modulación sea mucho menor que la anchura de la línea EPR (Fig. 7.3). Dado que la relación señal-ruido es proporcional a, uno suele medir en, donde la distorsión de la forma de línea es tolerable para casi todas las aplicaciones. El análisis preciso de la forma de línea puede requerir, mientras que la máxima sensibilidad a expensas del ensanchamiento artificial significativo de la línea se obtiene en. La frecuencia de modulación no debe ser más amplia que el ancho de línea en las unidades de frecuencia. Sin embargo, con la frecuencia de modulación estándar que corresponde en una escala de campo magnético a solo at, esto rara vez es un problema.
16.1.3. Consideraciones sobre la preparación de muestras
Dado que los espines electrónicos tienen un momento magnético mucho mayor que los espines nucleares, los acoplamientos electrón-electrón conducen a un ensanchamiento significativo de la línea en soluciones concentradas. Las concentraciones de los centros paramagnéticos generalmente no deben exceder para evitar dicha ampliación. Para los radicales orgánicos en solución líquida puede ser necesario diluir la muestra a fin de lograr la resolución final. Para los dopantes metálicos paramagnéticos en compuestos hospedadores diamagnéticos, en la mayoría de los sitios diamagnéticos deben ser sustituidos por centros paramagnéticos. Dichas concentraciones se pueden detectar fácilmente y con buena relación señal/ruido. Para la mayoría de las muestras, se pueden obtener buenos espectros hasta el rango en solución y hasta el rango de dopante de 100 ppm en sólidos.
El ensanchamiento de la línea en solución líquida también puede surgir de la colisión difusional de paramagnéticos

Figura 7.3: Detección de la forma de línea derivada por modulación de campo. La situación se considera en el campo instantáneo durante un barrido de campo (línea discontinua vertical) que es lento en comparación con la frecuencia de modulación de campo de. La modulación del campo magnético con amplitud (azul) provoca una modulación de la señal de salida (roja) con la misma frecuencia y una amplitud. La detección sensible a la fase mide esta amplitud, que es proporcional a la derivada del lineshape de absorción gris y a, siempre que sea mucho menor que el ancho de línea pico a pico de la línea. En la práctica, suele ser aceptable. Para un análisis preciso de la forma de línea, se recomienda.
Figura 7.4: Mejora de la relajación por intercambio de colisiones con oxígeno en solución. a) Situación previa al encuentro difusional. Como ejemplo, se supone que el oxígeno triplete está en un estado (rojo), mientras que se supone que el espín de un radical nitróxido es (verde). b) La molécula de oxígeno y el radical nitróxido han colisionado durante el encuentro difusional. Sus funciones de onda se superponen y los tres electrones desapareados no pueden distinguirse entre sí (gris). (c) Después de la separación, los tres electrones desapareados se han redistribuido arbitrariamente a las dos moléculas. Por ejemplo, ahora el oxígeno puede estar en el estado (rojo) y el nitróxido en el estado (verde). El espín electrónico del radical nitroxido se ha volteado.
especies con oxígeno triplete paramagnético (Figura 7.4). Durante tal colisión, las funciones de onda de las dos moléculas se superponen y, dado que los electrones son partículas indistinguibles, los estados de espín de todos los electrones desapareados en ambas moléculas se redistribuyen arbitrariamente cuando las dos moléculas se separan de nuevo. Los encuentros difusionales estocásticos conducen así a giros adicionales de los espines de electrones observados, lo que corresponde a la relajación y acorta el tiempo de relajación longitudinal. Dado que el ancho de línea es proporcional y no puede ser mayor que, los frecuentes encuentros colisionales de especies paramagnéticas conducen al ensanchamiento de la línea. Dicha ampliación de línea aumenta con la disminución de la viscosidad (difusión más rápida) y el aumento de la concentración de oxígeno El efecto es más fuerte en disolventes apolares, donde la solubilidad de oxígeno es mayor que en los disolventes polares, pero a menudo es significativo incluso en solución acuosa. La mejor resolución se obtiene si la muestra está libre de oxígeno. El mismo mecanismo conduce al ensanchamiento de líneas a alta concentración de una especie paramagnética en solución líquida. En estado sólido, el ensanchamiento de la línea a alta concentración se debe principalmente al acoplamiento dipolo-dipolo. A menudo, el espectro de EPR ampliado anisotrópicamente en estado sólido es de interés, ya que proporciona información sobre anisotropía y acoplamientos hiperfinos anisotrópicos. Esto puede requerir la congelación de una solución de la especie de interés. Por lo general, la especie precipitará si el disolvente cristaliza, lo que puede provocar el ensanchamiento de la línea y, en casos extremos, incluso el colapso de la estructura hiperfina y el promedio de anisotropía por intercambio entre especies paramagnéticas vecinas. Estos problemas se previenen si el disolvente forma un vidrio, como suele ser el caso de los disolventes que tienen grupos metilo o pueden formar enlaces de hidrógeno en geometrías muy diferentes. Los disolventes formadores de vidrio típicos son tolueno, 2-metiltetrahidrofurano, etanol, etilenglicol y glicerol. Las soluciones acuosas requieren la adición de al menos glicerol como crioprotector. En la mayoría de los casos, la cristalización seguirá ocurriendo con enfriamiento lento. Por lo tanto, las muestras se congelan por choque por inmersión del tubo de muestra en nitrógeno líquido. Los tubos de vidrio se romperían por inmersión directa en nitrógeno líquido, pero los espectros EPR tienen que medirse en tubos de muestra de sílice fundida de todos modos, ya que el vidrio contiene invariablemente una cantidad detectable de impurezas paramagnéticas de hierro.
16.2. Descripción teórica de CW EPR
Esta sección se solapa con la Sección de las notas de conferencias de RMN.
16.2.1. Paquete Spin Lineshape
Todos los giros en una muestra que tienen la misma frecuencia de resonancia forman un paquete de giro. A continuación también asumimos que todos los giros de un paquete de giro tienen los mismos tiempos de relajación longitudinal y transversal y, respectivamente. Si el número de giros en el paquete de giro es suficientemente grande, podemos asignar un vector de magnetización al paquete de giro. La dinámica de este vector de magnetización con magnetización de equilibrio durante la irradiación de microondas se describe mediante las ecuaciones de Bloch en el marco giratorio. En la espectroscopia EPR, es inusual utilizar la relación giromagnética. De ahí que denotemos la resonancia compensada por
donde está la frecuencia de microondas en unidades de frecuencia. Las ecuaciones de Bloch de marco rotativo para los tres componentes del vector de magnetización pueden escribirse como
donde es la amplitud del campo de microondas en unidades de frecuencia angular y es el valor medio en el plano perpendicular al campo magnético estático. La diferencia de signos aparente para los términos y surge del diferente sentido de precesión de espín para espines electrónicos en comparación con espines nucleares con una relación giromagnética positiva.
Si el paquete giratorio se irradia a frecuencia de microondas constante, potencia de microondas constante y campo magnético estático constante durante un tiempo suficientemente largo (aproximadamente), el vector de magnetización alcanza un estado estacionario. Aunque el campo estático es barrido en un experimento CW EPR, asumir un estado estacionario es una buena aproximación, ya que el barrido de campo suele ser lento en comparación con y. Los barridos más rápidos corresponden al régimen de escaneo rápido que no se trata en esta clase magistral. En el estado estacionario, los lados izquierdos de las ecuaciones diferenciales (7.2) para los componentes del vector de magnetización deben ser todos cero,
Este sistema lineal de ecuaciones tiene la solución
donde no suele detectarse, está en fase con la irradiación excitante de microondas y corresponde a la señal de dispersión, y está desfasada con la irradiación excitante y corresponde a la línea de absorción. Las formas de línea no perturbadas se obtienen en el régimen lineal, donde el parámetro de saturación
cumple. Se puede determinar fácilmente a partir de la Ec. (7.4) que en el régimen lineal aumenta linealmente con el aumento, lo que corresponde a la proporcionalidad de la señal a la raíz cuadrada de la potencia de microondas. está muy cerca de la magnetización de equilibrio. Dentro de este régimen, una disminución de la atenuación de microondas, es decir, un aumento de potencia en, aumenta la amplitud de la señal en un factor de 2. La forma de línea no depende en el régimen lineal. Por lo tanto, es una buena práctica medir a la mayor potencia de microondas que aún está bien dentro del régimen lineal, ya que esto corresponde a la máxima relación señal/ruido. Para mayor potencia la línea se ensancha.
Dentro del régimen lineal, toma la forma de una línea de absorción lorentziana
con ancho de línea en unidades de frecuencia angular. El ancho de línea pico a pico de la primera derivada de la línea de absorción es. Dado que los espectros EPR se miden por barrido de campo magnético, necesitamos convertir a unidades de campo magnético,
El ancho de línea de un paquete de giro se denomina ancho de línea homogéneo. Si es lo mismo para todos los paquetes spin, este ancho de línea homogéneo es proporcional a, hecho que debe tenerse en cuenta en las simulaciones de lineshape para sistemas con gran anisotropía. Para la mayoría de las muestras, el ensanchamiento adicional de la línea surge de acoplamientos hiperfinos no resueltos y, en estado sólido, anisotropía. Por lo tanto, generalmente no se puede obtener aplicando la Eq. (7.7) al ancho de línea pico a pico observado experimentalmente.
16.2.2. Saturación
Para una potencia de microondas mayor que en el régimen lineal, el ancho de línea pico a pico aumenta en un factor. Si es necesario detectar una señal débil con una relación señal/ruido máxima, es ventajoso aumentar la potencia más allá del régimen lineal, pero no necesariamente al nivel máximo disponible. Para irradiación muy fuerte, se puede descuidar el término 1 en el denominador de las ecuaciones (7.4) para los componentes del vector de magnetización. La amplitud de resonancia de la línea de absorción viene dada por
es decir, es inversamente proporcional a. En este régimen, la amplitud disminuye con el aumento de la potencia de la irradiación de microondas.
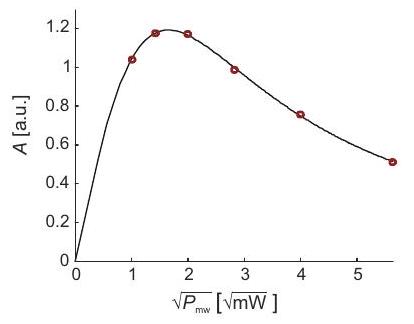
Figura 7.5: Medición progresiva de saturación en la proteína de membrana LHCII solubilizada en micelas detergentes en atmósfera de nitrógeno. El residuo V229 se mutó a cisteína y se marcó por espín mediante yodoacetamidoproxilo. Los puntos de datos experimentales (rojo) se obtuvieron a las atenuaciones de microondas de, y con una potencia total de. El ajuste por la Ec. (7.9) (línea negra) proporciona y.
La información semicuantitativa sobre la relajación de espín se puede obtener mediante el experimento de saturación de potencia progresiva, donde el espectro EPR se mide en función de la potencia de microondas. Por lo general, la amplitud pico a pico de la señal de grandes dimensiones en el espectro se representa como una función de. Tales curvas de saturación se pueden ajustar mediante la ecuación
donde el parámetro de inhomogeneidad toma el valor en el límite homogéneo y en el límite no homogéneo. Por lo general, no se conoce de antemano y se trata como un parámetro de ajuste. Los otros parámetros de ajuste son, que es la pendiente del aumento de amplitud con la raíz cuadrada de la potencia de microondas en el régimen lineal, y, que es la potencia de media saturación. Más precisamente, es el mw de potencia incidente donde se reduce a la mitad de su valor insaturado. La Figura muestra datos experimentales de una medición de saturación progresiva en el mutante V229C marcado con espín del complejo principal de recolección de luz vegetal LHCII solubilizado en micelas detergentes en una atmósfera de nitrógeno y un ajuste de estos datos por la Ec. (7.9).


16.3. ENDOR
16.3.1. Ventajas de la detección de espectros de frecuencia nuclear basada en espín electrónico
Los espectros de frecuencia nuclear en los estados líquido (Sección 4.3.2) y sólido (4.3.4) presentan una resolución hiperfina mucho mejor que los espectros EPR, debido a que los espectros anteriores presentan menos líneas y más estrechas. De hecho, los pequeños acoplamientos hiperfinos a núcleos de ligando en complejos metálicos no suelen resolverse en espectros EPR y solo los acoplamientos hiperfinos más grandes pueden resolverse en espectros EPR de estado sólido. Los espectros de frecuencia nuclear no se pueden medir con un espectrómetro de RMN dedicado porque se extienden sobre varios megahercios a varias decenas de megahercios, mientras que los espectrómetros de RMN están diseñados para anchos de banda de excitación y detección de algunas decenas de kilohercios. Además, las transiciones de espín electrónico tienen 660 veces más polarización que las transiciones de protones y más que la de otros núcleos. Su mayor momento magnético también conduce a una mayor sensibilidad de detección. Por lo tanto, es ventajoso transferir la polarización de espines de electrones a espines nucleares y retrotransferir la respuesta de los espines nucleares a los espines de electrones para su detección. Dos clases de experimentos pueden lograr esto, los experimentos de doble resonancia nuclear electrónica (ENDOR), discutidos en la Sección y los experimentos de modulación de envolvente de eco de espín electrónico (ESEEM) discutidos en la Sección 8.2
16.3.2. Tipos de experimentos ENDOR
Se puede realizar un experimento ENDOR con una fuerte irradiación CW de espines electrónicos y nucleares. En este experimento CW ENDOR, una transición de espín electrónico está parcialmente saturada, en la Ec. (7.5). Al conducir una transición de espín nuclear que comparte un nivel de energía con la transición saturada, se abren vías de relajación adicionales. La transición de espín electrónico bajo observación es así parcialmente desaturada y se observa un aumento en la señal EPR. El experimento se realiza en campo magnético constante con fuerte irradiación de microondas a un máximo del espectro de absorción de primera derivada (es decir, el espectro CW EPR) y la señal EPR se registra en función de la frecuencia de irradiación de radiofrecuencia adicional, que debe cumplir con la saturación condición para los giros nucleares. Por lo general, la irradiación por radiofrecuencia es modulada en frecuencia y la respuesta se detecta con otro detector sensible a la fase, lo que lleva a la observación de la primera derivada del espectro de frecuencias nucleares. El experimento CW ENDOR depende críticamente de un equilibrio de tiempos de relajación, por lo que en estado sólido solo se puede lograr una sensibilidad suficiente en un cierto rango de temperatura. Además, la irradiación continua fuerte simultánea tanto por microondas como por radiofrecuencia, mientras se mantiene constante la frecuencia y la temperatura del resonador, es experimentalmente desafiante. Por lo tanto, CW ENDOR ha sido reemplazado en gran medida por técnicas ENDOR pulsadas Sin embargo, para muestras de solución líquida CW ENDOR suele ser la única técnica ENDOR aplicable.
El experimento ENDOR pulsado conceptualmente más simple es Davies ENDOR (Sección 8.1.3), donde la saturación de la transición EPR es reemplazada por inversión por un pulso (Fig. 8.1 (a)). Un pulso de radiofrecuencia posterior, que es on-resonante con una transición que comparte un nivel con la transición EPR invertida, cambia la población de este nivel y así la polarización de la transición del observador EPR. Este cambio de polarización en función de la radiofrecuencia se observa mediante un experimento de eco de Hahn en la transición del observador. El enfoque funciona bien para acoplamientos hiperfinos moderadamente grandes, en particular para núcleos coordinados directamente con un ion de metal de transición o para protones a una distancia de enlace de hidrógeno o distancias de hasta aproximadamente Å. Como veremos en la Sección 8.1.3, el experimento es bastante insensible para acoplamientos hiperfinos muy pequeños.
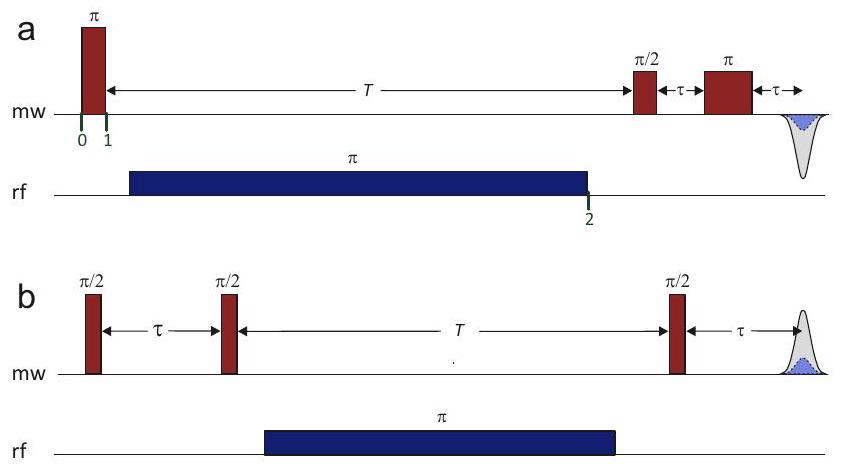
Figura 8.1: Secuencias ENDOR pulsadas. a) Davies ENDOR. Un pulso de inversión selectiva en los espines electrónicos es seguido por un retardo y detección de eco de Hahn (rojo). Durante el retardo interpulso de microondas, se aplica un pulso de radiofrecuencia variable en frecuencia (azul). Si este pulso está encendido resonante con una transición nuclear, el eco invertido se recupera (azul pálido). b) Mims ENDOR. Una secuencia de eco estimulada no selectiva con retardos interpulso y se aplica a los espines de electrones (rojo). Durante el retardo interpulso de microondas, se aplica un pulso de radiofrecuencia variable en frecuencia (azul). Si este pulso está encendido resonante con una transición nuclear, el eco estimulado se atenúa (azul pálido).
Los acoplamientos hiperfinos más pequeños se pueden detectar con el experimento Mims ENDOR que se basa en la secuencia de eco estimulado Fig. b)). El bloque de preparación crea una rejilla de polarización de la forma funcional, donde está el espectro de absorción EPR en función del desplazamiento de resonancia y es el retardo entre los dos pulsos de microondas. Se aplica un pulso de radiofrecuencia con frecuencia variable durante el tiempo en que la magnetización de espín electrónico se alinea con el eje. Si este pulso está encendido resonante con una transición nuclear que comparte un nivel con la transición EPR del observador, la mitad de la rejilla de polarización es desplazada por la división hiperfina, como también se hará evidente en la Sección 8.1.3. Porque con entero la rejilla de polarización es destruida por interferencia destructiva. Dado que el eco estimulado es el decaimiento de inducción libre (FID) de esta rejilla de polarización, es cancelado por un pulso de radiofrecuencia que está en resonancia con una transición nuclear. Es evidente que el pulso de radiofrecuencia no tiene efecto para con entero, donde las rejillas originales y de frecuencia desplazada interfieren constructivamente. Por lo tanto, el experimento Mims ENDOR presenta puntos ciegos en función del retardo interpulso. Estos puntos ciegos no son un problema grave para acoplamientos hiperfinos muy pequeños. Tenga en cuenta sin embargo que el primer punto ciego corresponde a. Por lo tanto, se requieren retardos interpulso largos para detectar acoplamientos hiperfinos muy pequeños, lo que conduce a una fuerte atenuación del eco por un factor debido a la relajación transversal del espín electrónico. Se puede demostrar que la máxima sensibilidad para acoplamientos muy pequeños se alcanza aproximadamente a.

Figura 8.2: Transferencia de polarización en Davies ENDOR. (a) Poblaciones niveladas en equilibrio térmico, correspondientes a la marca verde 0 en la Fig. 8.1 (a). Las transiciones electrónicas (rojo, rojo pálido) están mucho más polarizadas que las transiciones nucleares (azul, azul pálido). (b) Poblaciones niveladas después de un pulso selectivo de inversión de mw en resonancia con la transición (rojo oscuro), correspondiente al marcador verde 1 en la Fig. 8.1 (a). Se genera un estado de orden de dos espines, donde las dos transiciones de espín de electrones se polarizan con signo opuesto y lo mismo es cierto para las dos transiciones de espín nuclear. (c) Poblaciones niveladas después de un pulso de inversión selectiva de rf en resonancia con la transición (azul oscuro), correspondiente a la etiqueta verde 2 en la Fig. 8.1 (a). La transición del observador de espín electrónico ya no está invertida, sino saturada.
16.3.3. Davies ENDOR
El experimento Davies ENDOR se entiende más fácilmente al observar las transferencias de polarización. En equilibrio térmico, las transiciones de espín electrónico (rojo y rojo pálido) están mucho más polarizadas que las transiciones de espín nuclear (Fig. 8.2 (a)). Sus frecuencias difieren por una división hiperfina efectiva a un espín nuclear que es azul codificado por colores. El primer pulso de microondas es selectivo para la transición, es decir, tiene un ancho de banda de excitación que es menor que. En consecuencia, invierte solo una de las dos transiciones de espín electrónico. Suponemos que la transición (roja) está invertida y la transición (rojo pálido) no se invierte; el otro caso es análogo. Dicha inversión selectiva de transición conduce a un estado de orden de dos espines, donde todas las transiciones individuales en el sistema de dos espinas están polarizadas. Sin embargo, las dos transiciones de espín electrónico están polarizadas con signo opuesto y las dos transiciones nucleares también están polarizadas con signo opuesto (Fig. 8.2 (b)). Ahora se aplica un pulso de radiofrecuencia. Si este pulso no es resonante con una transición nuclear, el estado de orden de dos espines persiste y la transición de espín electrónico observador (rojo) aún está invertida. El pulso de radiofrecuencia también es selectivo para la transición. Ahora asumimos que este pulso invierte la transición (azul); el otro caso vuelve a ser análogo. Después de dicho pulso de radiofrecuencia resonante, las dos transiciones nucleares se polarizan con signo igual y las dos transiciones de espín electrónico se saturan sin polarización existente en ellas (Fig. 8.2 (c)). Después del pulso de radiofrecuencia se aplica una secuencia de eco Hahn de microondas resonante con la transición del observador (Fig. 8.1 (a)). Si el pulso de radiofrecuencia estaba apagado resonante (situación como en la Fig. 8.2 (b)), se observa un eco invertido. Si, por otro lado, el pulso de radiofrecuencia estaba en resonancia (situación como en la Fig. 8.2 (c)) no se observa eco. En la práctica, las transferencias de polarización no son completas y aún se observa un eco débil. Sin embargo, un pulso de radiofrecuencia resonante provoca cierta recuperación del eco invertido. Si se varía la radiofrecuencia, se observa recuperación del eco invertido en todas las frecuencias donde el pulso de radiofrecuencia es resonante con una transición nuclear.
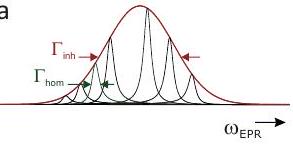
C


Figura 8.3: Explicación de quema de agujeros espectrales de Davies ENDOR. (a) Una línea EPR deshomogéneamente ensanchada con ancho (rojo) consiste en muchas líneas más estrechas homogéneamente ensanchadas con ancho de línea. (b) La irradiación de microondas débil larga satura el paquete de espín resonante y no afecta significativamente a los paquetes de espín fuera de resonancia. Un agujero espectral se quema en la línea deshomogéneamente ensanchada, que puede ser tan estrecha como. (c) Un pulso selectivo de microondas quema un orificio de inversión en la línea EPR cuyo ancho es aproximadamente el ancho inverso del pulso. d) Situación después de aplicar un pulso de radiofrecuencia resonante. Para el paquete de giro, donde el pulso de microondas estaba encendido-resonante con la transición, la mitad del agujero espectral se desplaza a frecuencias EPR más bajas. Para el paquete de giro donde el pulso de microondas estaba encendido-resonante con la transición, la mitad del agujero espectral se desplaza a frecuencias EPR más altas. Considerando ambos casos, la mitad del agujero persiste, correspondiente a la saturación. Se crean dos agujeros laterales con un cuarto de la profundidad del orificio de inversión en. Estos agujeros laterales no contribuyen a la señal de eco, siempre y cuando estén fuera de la ventana de detección (rojo pálido) cuyo ancho está determinado por el ancho de banda de excitación de la secuencia de detección de eco de Hahn.
Se obtiene una mayor comprensión de Davies ENDOR considerando una línea EPR no homogéneamente ensanchada (Fig. 8.3). En tal línea con ancho, cada paquete de giro individual con un ancho mucho más estrecho puede, en principio, excitarse selectivamente. Un pulso rectangular largo invierte el paquete de giro en resonancia y parcialmente invierte los paquetes de giro aproximadamente sobre un ancho de banda correspondiente a la longitud inversa del pulso. En Davies ENDOR, longitudes de pulso entre 50 y, correspondientes a anchos de banda de excitación entre 20 y son típicas. Tal pulso crea un orificio de inversión centrado en la frecuencia de microondas. En un sistema de espín electrón-nuclear, existen dos paquetes de espín on-resonante, aquellos donde está la frecuencia de la transición y aquellos donde es la frecuencia de la transición. Para el primer paquete de giro, la inversión del giro nuclear del al estado aumenta la frecuencia EPR por la división hiperfina efectiva, mientras que para el último paquete, la inversión del al estado lo disminuye en. En ambos casos, la mitad del agujero de inversión se desplaza a un orificio lateral, dejando un orificio de saturación y creando un orificio lateral de saturación. Los agujeros centrales de saturación de los dos paquetes de giro coinciden en frecuencia y se combinan con un agujero de saturación en la línea deshomogéneamente ensanchada. En las frecuencias de los orificios laterales, solo uno de los dos paquetes de giro contribuye al agujero, de manera que los orificios laterales son solo la mitad de profundos. La subsecuencia de eco de Hahn en la secuencia ENDOR de Davies debe tener un ancho de banda de detección que cubra solo el orificio central (rojo pálido en la Fig.), ya que no se observaría ningún efecto ENDOR si también se cubriera el orificio lateral. Para este propósito, el ancho de banda de detección de la secuencia de eco de Hahn se puede limitar ya sea usando pulsos de microondas suficientemente largos o usando una puerta de integración suficientemente larga para el eco invertido.
En cualquier caso, un efecto Davies ENDOR solo se observa si excede el ancho del agujero de inversión original. Cuanto más pequeño, más largo debe ser el primer pulso de inversión y menos paquetes de giro contribuyen a la señal. En general, no se pueden detectar divisiones hiperfinas mucho más pequeñas que el ancho de línea homogéneo en el espectro EPR. En la práctica, Davies ENDOR se vuelve muy insensible para longitudes de pulso superiores a 400 ns. Si el ensanchamiento del agujero de inversión por relajación del espín electrónico es insignificante, la supresión de señales con pequeños acoplamientos hiperfinos en Davies ENDOR puede describirse mediante un parámetro de selectividad
donde la longitud del primer pulso mw. Se obtiene la intensidad de ENDOR absoluta máxima para. En función de, la intensidad absoluta de ENDOR viene dada por
La selectividad de contraste hiperfina descrita por la Ec. (8.2) puede ser utilizada para la edición espectral. Por ejemplo, ENDOR señala átomos de nitrógeno del ligando directamente coordinados en complejos de metales de transición con del orden de superposición con señales ENDOR de protones de ligando débilmente acoplados a frecuencias de banda X. En una longitud de pulso de inversión de aproximadamente ENDOR las señales son suprimidas en gran medida.
La ventaja de sensibilidad de Mims ENDOR para acoplamientos hiperfinos muy pequeños también se puede entender en la imagen de quema de agujeros. En lugar de un solo orificio central, un bloque de preparación con pulsos de microondas no selectivos crea una rejilla de polarización que se puede imaginar como un peine de muchos agujeros que están espaciados por diferencia de frecuencia. El ancho de cada agujero es aproximadamente. El ancho del peine de agujeros está determinado por la longitud inversa de los pulsos no selectivos, que suelen ser largos. Para acoplamientos pequeños, donde en Davies ENDOR necesita ser muy largo, más de un orden de magnitud más paquetes de giro participan en un experimento Mims ENDOR que en un experimento de Davies ENDOR. El efecto Mims ENDOR surge del desplazamiento de una cuarta parte de la rejilla de polarización por diferencia de frecuencia y una cuarta parte de la rejilla por. Las rejillas desplazadas interfieren con la rejilla en la frecuencia central. Dependiendo de y de la periodicidad de la rejilla, esta interferencia es destructiva (efecto ENDOR) o constructiva (sin efecto ENDOR).
16.4. ESEEM e HYSCORE
16.4.1. ¿ENDOR o ESEEM?
En los experimentos de ESEEM, la transferencia de polarización de espines electrónicos a espines nucleares y la detección de frecuencias nucleares en transiciones de espín electrónico se basan en las transiciones electrón-nuclear prohibidas discutidas en el Capítulo 6. Gran parte de la mayor polarización de las transiciones de espín electrónico se pierde en tales experimentos, ya que el ángulo entre los ejes de cuantificación del espín nuclear en los dos estados de espín electrónico suele ser pequeño y la profundidad de las modulaciones de eco nuclear es pecado. Además, las modulaciones desaparecen a lo largo de los ejes principales del tensor hiperfino, donde y así. Por lo tanto, faltan singularidades de la forma de línea en los espectros unidimensionales de ESEEM, lo que complica significativamente el análisis de lineshape. Por esta razón, los experimentos unidimensionales de ESEEM no suelen ser competitivos con los experimentos ENDOR, al menos si los experimentos ENDOR pueden realizarse a frecuencias de banda o incluso frecuencias más altas. Una excepción surge para núcleos “remotos” débilmente acoplados en complejos de metales de transición donde se puede lograr la cancelación exacta entre las interacciones nuclear Zeeman e hiperfinas para uno de los estados de espín electrónico a frecuencias de banda X o ligeramente por debajo. En esta situación, se observan frecuencias cuadrupolares nucleares puras, lo que conduce a líneas estrechas y espectros fácilmente interpretables. Los datos unidimensionales de ESEEM también son útiles para determinar concentraciones locales de protones o deuterio alrededor de una etiqueta de espín, que puede ser utilizada como un proxy de accesibilidad al agua (Sección 10.1.6).
La principal ventaja de ESEEM en comparación con la espectroscopia ENDOR es la extensión más fácil de ESEEM a un experimento de correlación bidimensional. La espectroscopia de correlación subnivel hiperfina (HYSCORE) 8.2.3 resuelve la señal superpuesta de diferentes elementos, simplifica la asignación de picos y permite la determinación directa de la anisotropía del tensor hiperfino aunque no se observen las singularidades de la forma de línea.
16.4.2. ESEEM de tres pulsos
El experimento HYSCORE es una extensión bidimensional del experimento ESEEM de tres pulsos que trataremos primero. En este experimento, la amplitud de un eco estimulado después se observa con la secuencia de pulsos como una función del retardo interpulso variable en el retardo interpulso fijo (Fig. 8.4). El bloque sirve como generador de coherencia nuclear, como se discute en la Sección 6.3.1 y, simultáneamente, crea la rejilla de polarización discutida en el contexto del experimento Mims ENDOR (Sección 8.1.2). De hecho, la mayor parte de la magnetización de equilibrio térmico se convierte en la rejilla de polarización cuyo FID después del pulso final es el eco estimulado, mientras que sólo una pequeña fracción se transfiere a la coherencia nuclear. La fase de la coherencia nuclear determina cuánto contribuye al eco estimulado después de la retrotransferencia a la coherencia de espín electrónico por el último pulso. Para un sistema de espín electrón-nuclear esta fase evoluciona con frecuencias o si durante el retardo interpulso el espín electrónico está en su estado o, respectivamente. De ahí que la parte del eco estimulado que surge de la coherencia nuclear retrotransferida se modula en función de con frecuencias y.
Una expresión para la modulación de envolvente de eco se puede derivar por formalismo de operador de producto utilizando los conceptos explicados en la Sección 6.2. Sin tener en cuenta la relajación, la derivación algo larga proporciona
donde los términos y corresponden a contribuciones con el espín electrónico en su estado o, respectivamente, durante el retardo interpulso. Estos términos son dados por
Los factores para el término y para el término describen el comportamiento de punto ciego de ESEEM de tres pulsos. La profundidad de modulación viene dada por
Para pequeños acoplamientos hiperfinos,, tenemos, de manera que la Eq. (8.5) se reduce a
es decir, la profundidad de modulación es inversamente proporcional al cuadrado del campo magnético. Usando las ecuaciones (4.10) y (4.11) encontramos para protones no muy cercanos a un electrón desapareado bien localizado
donde está el ángulo entre el eje electrón-protón y el campo magnético estático.
Debido a la topología en estrella de los sistemas de espín electrón-nuclear (Fig. 4.4 (a)), la ecuación (8.3) se puede extender fácilmente mediante una regla de producto a múltiples núcleos con espines, donde es un índice que recorre todos los núcleos. Uno encuentra
En el límite de modulación débil, donde todas las profundidades de modulación cumplen la condición, el espectro ESEEM debido a varios núcleos acoplados es la suma de los espectros de los núcleos individuales.
a
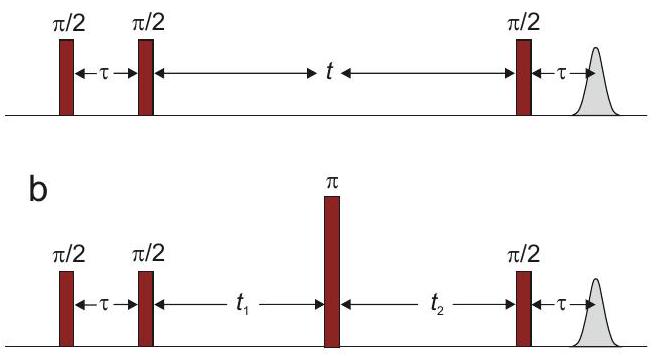
Figura 8.4: Secuencias de pulsos para ESEEM de tres pulsos (a) e HYSCORE (b). En ESEEM de tres pulsos, el tiempo es variado y el tiempo es fijo. En HYSCORE, los tiempos y se varían de forma independiente para obtener un conjunto de datos bidimensionales.
16.4.3. HYSCORE
El experimento HYSCORE se deriva del experimento ESEEM de tres pulsos insertando un pulso de microondas a mitad de camino a través de la evolución de la coherencia nuclear. Esto divide el retardo interpulso en dos retardos interpulso y (Fig. 8.4 (b)), los cuales se varían independientemente para proporcionar un conjunto de datos bidimensionales que depende paramétricamente del retardo interpulso fijo. El pulso insertado invierte el estado de espín electrónico. De ahí que la coherencia que ha evolucionado con la frecuencia durante el retardo interpulso evoluciona con la frecuencia durante el retardo interpulso y viceversa. En el límite de modulación débil, el experimento HYSCORE correlaciona solo frecuencias y del mismo espín nuclear. La expresión de modulación completa para el experimento HYSCORE contiene una contribución constante y contribuciones que varían solo con respecto a cualquiera o. Estas contribuciones se pueden eliminar mediante la corrección de fondo con funciones polinómicas de orden bajo a lo largo de ambas dimensiones. La modulación restante corresponde solo a picos cruzados y se puede expresar como
con
En esta representación con frecuencias nucleares sin signo, se tiene para el caso de acoplamiento débil y para el caso de acoplamiento fuerte, como se puede deducir de la Fig. 6.1. De ahí que en el caso de acoplamiento débil y en el caso de acoplamiento fuerte. En el caso de acoplamiento débil, los picos cruzados que correlacionan frecuencias nucleares con el mismo signo (términos) son mucho más fuertes que aquellos que correlacionan frecuencias con términos opuestos) mientras que es al revés en el caso del acoplamiento fuerte. Por lo tanto, los dos casos pueden distinguirse fácilmente en espectros HYSCORE, ya que los picos cruzados aparecen en diferentes cuadrantes (Fig. 8.5). Además, sin tener en cuenta un pequeño desplazamiento que surge de la parte pseudo-secular del acoplamiento hiperfino (ver más abajo), los picos cruzados de un isótopo dado con espín se sitúan en paralelos a la antidiagonal que corresponde a la frecuencia nuclear de Zeeman. Esta frecuencia a su vez se puede calcular a partir del valor nuclear (o relación giromagnética) y el campo magnético estático. Por lo tanto, la asignación de picos para los núcleos es sencilla. Para los núcleos con los picos se dividen aún más por la interacción del cuadrupolo nuclear. A menos que esta división sea mucho menor que la interacción hiperfina y la interacción nuclear de Zeeman, se requieren simulaciones numéricas para asignar los picos y extraer el acoplamiento hiperfino y cuadrupolo nuclear.
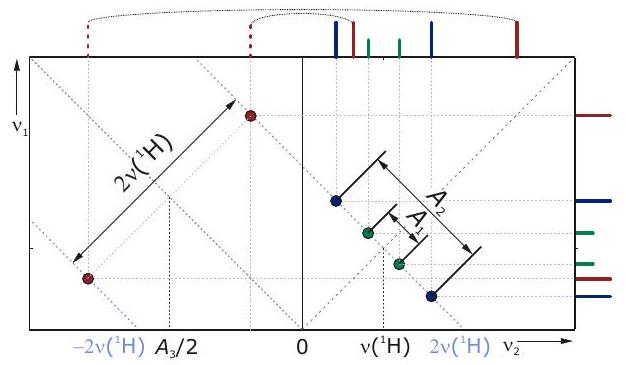
Figura 8.5: Espectro HYSCORE esquemático para el radical fenilo (compárese la Fig. 4.6). Tenga en cuenta que los acoplamientos hiperfinos se dan aquí en unidades de frecuencia, no en unidades de frecuencia angular. Las señales de núcleos débilmente acoplados aparecen en el cuadrante derecho. En primer orden, estos picos se sitúan en una línea paralela a la antidiagonal que cruza el eje en. Los dobletes están centrados y divididos por los respectivos acoplamientos hiperfinos. Las señales de núcleos fuertemente acoplados aparecen en el cuadrante (-, +). En primer orden, estos picos se sitúan en dos líneas paralelas a la antidiagonal que intersectan el eje en y. Los dobletes están centrados en la mitad del acoplamiento hiperfino y divididos por.
El pequeño desplazamiento pseudo-secular de los picos de correlación con respecto a la antidiagonal contiene información sobre la anisotropía de la interacción hiperfina (Fig. 8.5). En estado sólido, los picos cruzados de diferentes orientaciones forman crestas curvas. Para un tensor hiperfino con simetría axial, como se encuentra para protones no muy cercanos a un electrón desapareado bien localizado, el desplazamiento máximo en la dirección diagonal corresponde y viene dado por. Ya que se conoce,, y así la distancia electrón-protón se puede calcular a partir de este desplazamiento máximo. Si, que suele ser el caso, la orientación con desplazamiento máximo es al mismo tiempo la orientación con profundidad de modulación máxima.
Las crestas curvas terminan en su intersección con la paralela a la antidiagonal. Estos puntos corresponden a los valores principales del tensor hiperfino y la profundidad de modulación es cero en estos puntos. Sin embargo, generalmente es posible ajustar la cresta teórica a la cresta observada experimentalmente, ya que la curvatura cercana junto con la posición del punto determina completamente el problema.
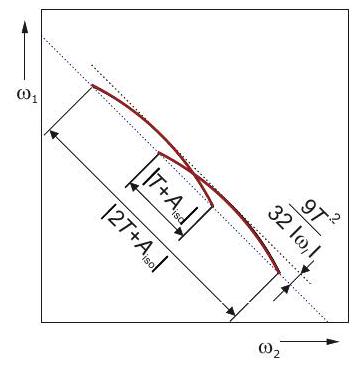
Figura 8.6: Espectro HYSCORE esquemático para un protón con un tensor hiperfino axial con anisotropía y componente isotrópico. Los picos de correlación de diferentes orientaciones forman crestas curvas (rojas). La curvatura es más fuerte cuanto mayor es la anisotropía y la relación de anisotropía cuadrada a la frecuencia nuclear de Zeeman determina el desplazamiento máximo con respecto a la antidiagonal.
El análisis de los espectros HYSCORE requiere cierta precaución debido al comportamiento de punto ciego (factor en la ecuación (8.9)) y debido a la selección de orientación por el ancho de banda limitado de los pulsos de microondas que es mucho menor que el ancho espectral para los complejos de metales de transición. Por lo tanto, es prudente medir los espectros HYSCORE en varios valores de y en varias posiciones de observador dentro del espectro EPR.


A una distancia de entre dos electrones no apareados localizados, la división entre los “cuernos” del patrón Pake es aproximadamente para dos espines de electrones. Incluso los espectros EPR fuertemente inhomogéneamente ampliados generalmente tienen características más estrechas que eso (aproximadamente en un barrido de campo magnético). Dependiendo del ancho de las características más estrechas en el espectro EPR y de la disponibilidad de un espectro experimental o espectro simulado realista en ausencia de acoplamiento dipolo-dipolo, las distancias hasta pueden estimarse a partir del ensanchamiento dipolar mediante análisis de líneas. A distancias inferiores, dicho análisis se vuelve incierto debido a la contribución del acoplamiento de intercambio entre los dos espines electrónicos, que no pueden ser calculados por los primeros principios y no pueden predecirse con suficiente precisión mediante cálculos cuántico-químicos. Si los dos electrones desapareados están unidos por una cadena continua de enlaces conjugados, el acoplamiento de intercambio puede ser significativo hasta distancias mucho más largas.
Las mediciones de distancia son más valiosas para etiquetas de espín o centros paramagnéticos nativos en biomoléculas o macromoléculas sintéticas y ensamblajes supramoleculares. En tales sistemas, si los dos electrones desapareados no están unidos por un sistema -electrón, el acoplamiento de intercambio es despreciable con respecto al acoplamiento dipolo-dipolo para distancias mayores que. Tales sistemas a menudo pueden asumir diferentes conformaciones moleculares, es decir, su estructura no está perfectamente definida. Por lo tanto, la caracterización estructural se beneficia fuertemente de la posibilidad de medir distribuciones de distancia en escalas de longitud que son comparables a la dimensión de estos sistemas. Esta dimensión es del orden de 2 a, correspondiente a entre y. Para inferir la distribución de la distancia, este pequeño acoplamiento dipolo-dipolo necesita ser separado de interacciones anisotrópicas más grandes.
Esta separación de interacciones es posible observando el cambio de frecuencia de resonancia para un giro en un par (azul en la Fig. 5.3) que se induce al voltear el espín de su compañero de acoplamiento (rojo). En la Fig. La frecuencia de resonancia del giro del observador antes del volteo de su compañero de acoplamiento se indica mediante una línea discontinua. Si el compañero de acoplamiento está en su estado antes del giro (panel izquierdo en la figura 5.3), el campo local en el giro del observador aumentará al voltear el compañero de acoplamiento. Esto provoca un aumento de la frecuencia de resonancia del espín del observador por el acoplamiento dipolo-dipolo (ver Ec. (5.16)). Si el compañero de acoplamiento está en su estado antes del giro (panel derecho en la figura 5.3), el campo local en el giro del observador disminuirá al voltear el compañero de acoplamiento. Esto provoca una disminución de la frecuencia de resonancia del giro del observador por el acoplamiento dipolo-dipolo. En la aproximación a altas temperaturas, ambos casos tienen la
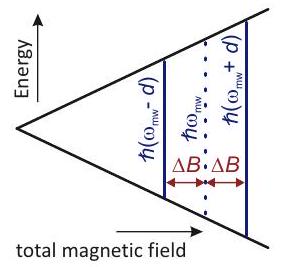
Figura 9.1: Desplazamiento de frecuencia de resonancia de un giro de observador (transiciones azules) por el cambio en el campo magnético local que surge al girar un segundo giro que es dipolo-dipolo acoplado al giro del observador. Compare la Fig. para la imagen de campo local.
misma probabilidad. De ahí que la mitad de los giros de los observadores experimentarán un cambio de frecuencia y la otra mitad experimentará un cambio de frecuencia. Si el giro del observador evoluciona con frecuencia cambiada durante algún tiempo, se ganarán fases en comparación con la situación sin voltear el compañero de acoplamiento. La fase adicional se puede observar como una modulación coseno para ambos casos, ya que el coseno es una función par.
16.5. VENADO
16.5.1. El experimento DEER de cuatro pulsos
El experimento más utilizado para las mediciones de distribución de distancia en el rango nanométrico es el experimento de resonancia electrónica doble de cuatro pulsos (DEER) (Figura 9.2), que a veces también se conoce como experimento de doble resonancia de electrones pulsados (PELDOR). Todas las interacciones del espín del observador se reenfocan dos veces por dos pulsos a veces y después del pulso inicial. El reenfoque repetido es necesario ya que todos los paquetes de giro deben estar en fase y la superposición del pulso de la bomba con el pulso del observador conduciría a la distorsión de la señal. El primer reenfoque con retardo interpulso restaura la situación (1) inmediatamente después del pulso con fase, donde los vectores de magnetización de todos los paquetes de giro se alinean con el eje. En la práctica, la coherencia se excita en ambas transiciones de giro del observador (azul en los paneles de nivel de energía), pero para mayor claridad consideramos solo la coherencia del giro del observador que está en la transición superior y está simbolizada por una línea ondulada en el panel (1).
Durante el tiempo posterior al primer reenfoque, los vectores de magnetización de paquetes de giro con diferente compensación de resonancia desfasan (panel (2)). Solo el paquete de giro en resonancia, marcado en azul oscuro, todavía está alineado con la dirección. El pulso de la bomba voltea el compañero de acoplamiento y, por lo tanto, transfiere la coherencia a la transición de giro inferior del observador La frecuencia de resonancia de esta transición es desplazada por el acoplamiento dipolo-dipolo en todos los paquetes de giro. La magnetización de giro del observador se desfase aún más hasta el momento justo antes de la aplicación del segundo pulso de observador (3)) y, además, todo el paquete de vectores de magnetización de paquetes de espín precede en sentido contrario a las agujas del reloj con el desplazamiento de frecuencia. Por lo tanto, el paquete de espín originalmente en resonancia gana fase antes de que se aplique el segundo pulso de observador. El segundo pulso de observador con fase corresponde a una rotación alrededor del eje. Esto refleja el haz de vectores de magnetización con respecto al eje, invirtiendo la fase de la coherencia de giro del observador (panel (4)). El paquete, que todavía
el primer reenfoque y todos los siguientes paneles de magnetización se reflejan con respecto al eje.
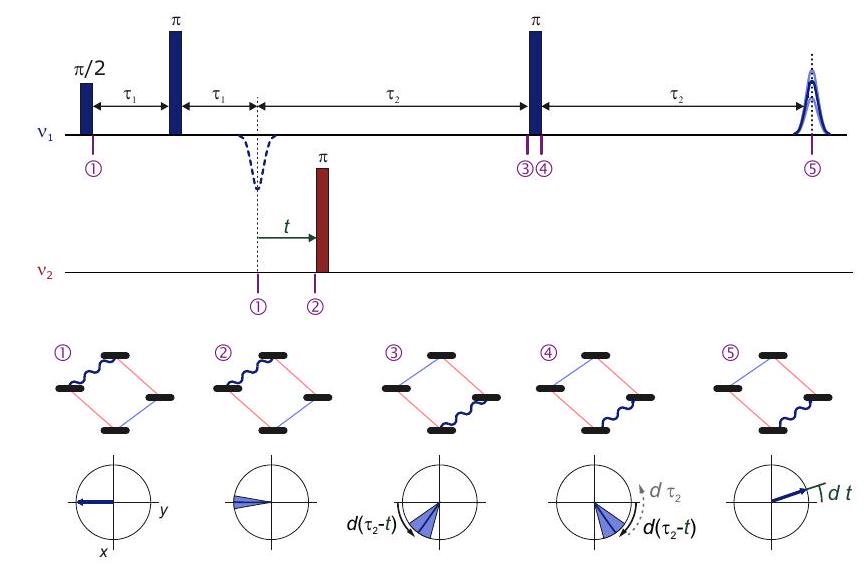
Figura 9.2: Secuencia DEER de cuatro pulsos, transferencias de coherencia y evolución de la magnetización de espín observador. Los pulsos mostrados en azul se aplican al giro del observador, el pulso de la bomba que se muestra en rojo se aplica a su compañero de acoplamiento. No se observa el eco en el tiempo (línea azul discontinua). Interpulse retarda y son fijos, el tiempo se varía, y la amplitud del eco se observa en función de.
precede en sentido contrario a las agujas del reloj con frecuencia angular ahora retarda el eje por fase. Durante el retardo interpulso final de longitud, el haz como un conjunto gana fase (flecha gris en el panel (4) y simultáneamente se vuelve a alinear a lo largo de su centro debido al reenfoque de eco. Sin embargo, el centro correspondiente al paquete de giro originalmente en resonancia no termina a lo largo, como lo habría hecho en ausencia del pulso de la bomba. Más bien, este paquete de giro ha ganado fase con respecto a la dirección (panel (5)). El componente del vector de magnetización a lo largo, que corresponde a la señal de eco, viene dado por.
El rango de distancia del experimento DEER está limitado hacia distancias cortas por el requisito de que, para el reenfoque de eco, los pulsos del observador deben excitar ambas transiciones del observador, que se dividen por y, para la transferencia de coherencia, el pulso de la bomba debe excitar ambas transiciones del compañero de acoplamiento, que también son dividido por. En otras palabras, tanto la subsecuencia de eco reenfocada del observador como el pulso de bombeo deben tener un ancho de banda de excitación que exceda. Este requisito establece un límite de distancia más bajo de aproximadamente a frecuencias de banda X y de aproximadamente a frecuencias de banda. Surge un límite hacia largas distancias, ya que es necesario observar varias oscilaciones dipolares para inferir el ancho o incluso la forma de una distribución de distancia y al menos se necesita observar una oscilación para determinar la distancia media. Esto requiere. Por otro lado, tenemos y el retardo interpulso fijo no puede ser mucho mayor que el tiempo de relajación transversal, ya que de lo contrario la coherencia se ha relajado completamente y no se observan ecos. Los tiempos de relajación transversal del espín electrónico son del orden de unos pocos microsegundos. Dependiendo del tipo de muestra (ver Sección), se puede elegir entre y, correspondiente a distancias máximas observables entre 5 y.
16.5.2. Requerimientos de muestra
En la vía de transferencia de coherencia deseada del experimento DEER, los pulsos de observador excitan exclusivamente los giros del observador y el pulso de la bomba excita exclusivamente al compañero de acoplamiento. El ancho de banda de excitación debe ser suficientemente grande para cubrir el acoplamiento dipolo-dipolo en todas las orientaciones, es decir, mayor que. Si los dos espines acoplados tienen el mismo espectro EPR, este espectro debe ser más amplio que el doble de este ancho de banda de excitación mínimo. Esta condición se cumple para las etiquetas de espín de nitróxido (Capítulo 10) y los iones de metales de transición en todas las frecuencias EPR, mientras que algunos radicales orgánicos, como los radicales tritilo, tienen espectros que son demasiado estrechos a frecuencias de banda X o incluso banda Q. Además, debe ser lo suficientemente largo para que al menos el observador gire. Esta condición puede cumplirse para casi todas las especies a temperaturas de (complejos de metales de transición) o (radicales orgánicos), pero puede requerir enfriamiento por debajo para algunas especies de alto espín. Para especies de espín alto con caparazón de valencia medio lleno, como Mn (II) o temperaturas de medición de también son suficientes.
La concentración de la muestra debe ser suficientemente baja para que las distancias intermoleculares sean mucho más largas que las distancias intramoleculares. Para distancias cortas son posibles concentraciones de hasta, pero las concentraciones de proporcionan mejores resultados, si se dispone de un espectrómetro con suficiente sensibilidad. Dependiendo de la distancia y, las mediciones se pueden realizar hasta concentraciones de. Para las proteínas de membrana reconstituidas en liposomas, la calidad de los datos no es solo una función de la concentración de espín en masa, sino también de la relación lípido-proteína. Este parámetro necesita ser optimizado para cada nueva proteína. El volumen de muestra requerido varía entre unos pocos microlitros (frecuencias de banda W) y siendo las frecuencias en banda Q generalmente óptimas.
Si la concentración no es demasiado alta y se puede alcanzar el límite de baja temperatura de relajación transversal, depende de la concentración y tipo de protones alrededor del giro del observador. La deuteración del solvente y el crioprotector (generalmente glicerol) generalmente mejoran drásticamente la calidad de los datos. Si la matriz puede ser perdeuterada, la deuteración de la proteína o el ácido nucleico puede prolongar y extender el rango de distancia o mejorar la relación señal/ruido.
Las complicaciones surgen si se encuentran más de dos electrones desapareados en una misma molécula, pero estas complicaciones suelen resolverse. Sin embargo, ninguno de los pares de espín debe tener una distancia más corta que el límite inferior del rango de distancia accesible.
17. Conversión de datos de evolución dipolar a distribuciones de distancia
17.5.3. Expresión para la señal DEER
En la Sección 9.1.1 hemos visto que el eco se modula con. Por lo general, esto se aplica solo a una fracción del eco, porque el pulso de la bomba excita solo una fracción de todos los paquetes de espín del compañero de acoplamiento del giro del observador. Por lo tanto, la señal de eco para un par aislado de espines de electrones en una orientación fija con respecto al campo magnético se describe mediante
donde la dependencia viene dada por las ecuaciones (5.16) y (5.15). La dependencia no puede expresarse en forma cerrada, pero a menudo se correlaciona tan débilmente con la que puede asumirse como un parámetro empírico constante. En esta situación, la ecuación (9.1) se puede integrar sobre todas las orientaciones
El pulso de la bomba invierte no solo el compañero de acoplamiento del espín observador en la misma molécula, sino también los espines electrónicos en otras moléculas remotas. Si estos giros vecinos se distribuyen homogéneamente en el espacio, el factor de fondo que surge de ellos asume la forma
donde la eficiencia de inversión promediada por orientación es la fracción de espines excitados por el pulso de la bomba, es un valor promedio y es la concentración total de espines. Por razones sutiles, difiere significativamente de la profundidad empírica de modulación de dos espines. Las distribuciones homogéneas de espines vecinos que están casi confinados a un plano o una línea dan lugar a una función de fondo exponencial estirada, donde es una dimensión fraccionaria de la distribución que suele ser algo mayor que 2 o 1 para distribuciones casi planas o lineales, respectivamente. La señal total de DEER viene dada por
Si la distancia se distribuye con densidad de probabilidad normalizada, el factor de forma necesita ser reemplazado por.
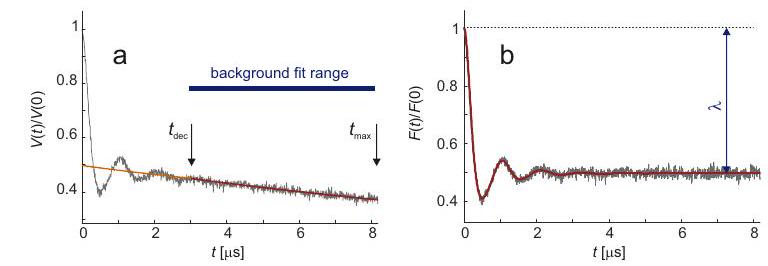
Figura 9.3: Corrección de fondo en espectroscopia DEER. (a) Datos primarios (simulación) normalizados a. La modulación dipolar decae hasta cierto tiempo. Una función de decaimiento exponencial (rojo) se ajusta a los datos en el rango, donde es el tiempo máximo de evolución dipolar. Esta función de fondo es extrapolada al rango (ocre). b) El factor de forma se obtiene normalizando la función de fondo, y dividiendo los datos primarios normalizados por. Decae a un nivel constante, donde está la profundidad de modulación. La curva roja es una simulación correspondiente a la distribución de distancia extraída por la regularización de Tikhonov con parámetro de regularización óptima.
17.5.4. Corrección de fondo
La información sobre la distribución de la distancia está contenida en, de la cual, por lo tanto, debe separarse de. A menudo, la distribución es suficientemente amplia para que las oscilaciones dipolares desaparezcan en un tiempo más corto que el tiempo máximo de evolución dipolar (Fig. 9.3 (a)). Para, la señal primaria es dada entonces por más ruido. La expresión for se ajusta a los datos primarios en este rango (línea roja en la Fig. 9.3 (a)). En algunos casos, por ejemplo para las proteínas solubles, se puede suponer una distribución homogénea de moléculas en tres dimensiones, de manera que se puede fijar. De lo contrario, se trata como un parámetro de ajuste, como son y. La función de fondo se obtiene extrapolando al rango (línea ocre) y dividiéndola por. De acuerdo con la Ec. (9.4), el factor de forma resulta dividiendo por. Para distribuciones de distancias estrechas, las oscilaciones en pueden durar hasta el más largo alcanzable. Esto no crea un problema si al menos la primera oscilación se completa mucho antes. Todas las siguientes oscilaciones tienen una amplitud muy similar y no sesgan el ajuste de fondo. Como regla general, se puede obtener una buena estimación para ajustando los datos a if, es decir, si se pueden observar dos oscilaciones completas. Si el rastreo de datos es más corto que eso, el ajuste de fondo está plagado de incertidumbre. La corrección de fondo incorrecta puede suprimir largas distancias o crear picos artificiales a largas distancias.
17.5.5. Regularización de Tikhonov con restricción de no negatividad
Para extraer la distribución de distancia del factor de forma experimental, necesitamos eliminar la contribución constante y renormalizar a la función de evolución dipolar
e invertir la ecuación integral, donde el núcleo viene dado por
Aquí, hemos sustituido por por e invertido dirección de la integración, que compensó el negativo en.
En la práctica, se digitaliza y se da como vector en tiempos de muestreo. De igual manera, es suficiente calcular como vector a distancias de muestreo. La ecuación integral se transforma así en una ecuación matricial
Desafortunadamente, esta ecuación matricial no se puede invertir fácilmente, ya que las filas del kernel no son ortogonales, es decir, el producto escalar de los vectores de función de evolución dipolar en diferentes no es cero. La débil dependencia lineal de las filas hace que el problema esté mal planteado. Pequeñas desviaciones de lo experimental de lo “verdadero”, por ejemplo debido al ruido, causan grandes desviaciones de la distribución de distancia verdadera. Este problema sólo puede resolverse tomando en cuenta información adicional.
Primero, lo sabemos, como densidad de probabilidad, en absoluto. De ahí que podamos imponer una restricción de no negatividad sobre. Resulta que esto no es suficiente para estabilizar la solución. El ruido se puede ajustar mediante distribuciones de distancias desiguales con muchos picos estrechos, aunque sabemos que la distribución de distancia debe ser suave, ya que surge de una distribución continua de conformaciones moleculares. La regularización de Tikhonov impone una restricción de suavidad al minimizar
donde
es la desviación cuadrática media entre datos experimentales y simulados y
es la norma cuadrada de la segunda derivada, que puede calcularse a partir de multiplicar con el operador de la segunda derivada. El parámetro de regularización determina el

Figura 9.4: Regularización Tikhonov de los datos mostrados en la Fig. 9.3. (a) Curva L. El parámetro de regularización óptima corresponde a la esquina (círculo verde) y proporciona la simulación mostrada en la Fig. 9.3 (b) así como la distribución de distancia extraída mostrada como una línea negra en el panel (c) de la Figura actual. El círculo rojo marca un parámetro de regularización demasiado grande que conduce a un sobrealisamiento. (b) Factor de forma de entrada (negro) y simulación para el parámetro de regularización demasiado grande correspondiente al círculo rojo en la curva. (c) Distribución teórica de distancia utilizada para simular un factor de forma sin ruido (verde) y distribución de distancia extraída del factor de forma ruidoso con parámetro de regularización óptimo correspondiente al círculo verde en la curva (negro). (d) (c) Distribución teórica de distancia utilizada para simular un factor de forma sin ruido (verde) y distribución de distancia extraída del factor de forma ruidoso con un parámetro de regularización demasiado grande correspondiente al círculo rojo en la curva (negro).
peso de la restricción de suavizado con respecto a la desviación cuadrática media entre datos experimentales y simulados. Una gráfica paramétrica de versus en función de es aproximadamente en forma de L (Fig. 9.4). Para muy pequeños, la rugosidad de la distribución de la distancia se puede disminuir fuertemente sin aumentar mucho la desviación media cuadrada. Para grandes ya es suave y un aumento adicional de conducirá sólo a una pequeña disminución de la rugosidad, sino a un gran aumento en, ya que la distribución de distancia demasiado ensanchada ya no se ajusta a las oscilaciones dipolares. De ahí que, en un sentido matemático, el parámetro de regularización óptima corresponda a la esquina de la curva. En este parámetro de regularización la distribución de distancia extraída (línea negra en la Fig. 9.4 (c)) es solo ligeramente más ancha que la distribución de distancia verdadera (línea verde) y el factor de forma simulado (línea roja en la Fig. 9.3 (b)) concuerda con el factor de forma experimental (línea negra), excepto el ruido blanco contribución. Si el parámetro de regularización es demasiado grande (círculo rojo en la Fig. 9.4 (a)), el factor de forma simulado se sobrehumedece (línea roja en la Fig. 9.4 (b)) y la distribución de distancia es irrealista amplia (línea negra en la Fig. 9.4 (d)). Para un parámetro de regularización demasiado pequeño, la distribución de distancia se divide de manera poco realista en varios picos estrechos y el factor de forma simulado se ajusta a parte del ruido (no mostrado). Este error no se puede discernir tan claramente en el factor de forma simulado como el sobreamortiguamiento se puede discernir. El subalisamiento es evidente solo en la curva L.

Sondas de centrifugado y etiquetas de nitroxido Sondas de centrifugado
Radicales Nitróxido
Espectro de EPR de Nitróxido Influencia de la dinámica en el espectro de nitroxido Polaridad y proticidad
Accesibilidad al agua
Accesibilidad al oxígeno Mediciones locales de pH
Trampas giradas
18. 0 - Sondas de centrifugado y trampas de centrifugado
18.1. Sondas de centrifugado y etiquetas de nitroxido
18.1.1. Sondas giratorias y etiquetas
Las sondas de espín son especies paramagnéticas estables que se mezclan a una muestra para obtener información estructural o dinámica sobre su entorno y, por lo tanto, indirectamente sobre la muestra. Las etiquetas de espín son sondas de espín que están unidas covalentemente a una molécula de interés, a menudo en un sitio específico. En comparación con la caracterización más directa de la estructura y la dinámica por otras técnicas, la espectroscopia EPR en sondas de espín puede ser capaz de acceder a otras escalas de longitud y tiempo o puede ser aplicable en estados de agregación o entornos donde estas otras técnicas exhiben baja resolución o no producen ninguna señal. El etiquetado de espín dirigido al sitio (SDSL) tiene la ventaja de que ya se conoce la asignación de la señal a la estructura molecular primaria y que un sitio específico en un sistema complejo puede estudiarse sin perturbación de las señales de otras partes del sistema. Este enfoque se beneficia de la rareza de los centros paramagnéticos. Por ejemplo, muchas proteínas y la mayoría de los ácidos nucleicos y lípidos son diamagnéticos. Si se introduce una etiqueta giratoria en un sitio seleccionado, la información de EPR es específica de este sitio en particular.
En principio, cualquier especie paramagnética estable puede servir como sonda de espín. Algunos iones metálicos paramagnéticos pueden sustituir a los iones diamagnéticos nativos del sistema en estudio, ya que tienen carga y radio iónico similares o con propiedades de complejación similares a las de los iones nativos. Esto se aplica a, que a menudo puede sustituir sin afectar la función de proteínas o ácidos nucleicos, o iones lantánidos Ln (III), que se unen a sitios Ca (II). Los iones metálicos paramagnéticos también pueden unirse a proteínas mediante ingeniería genética de sitios de unión con aminoácidos coordinantes, tales como histidina, o mediante la unión dirigida al sitio de un ligando metálico a la biomolécula. Dichos enfoques se utilizan para iones lantánidos, en particular Gd (III) y Cu (II).
Para muchos enfoques de sonda de espín, los radicales orgánicos son más adecuados que los iones metálicos, ya que en los radicales el electrón desapareado tiene un contacto más cercano con su entorno (los ligandos filtran el acceso ambiental de iones metálicos, en particular para iones lantánidos) y los espectros EPR son más estrechos, lo que permite algunos experimentos que no se pueden realizar en especies con espectros muy amplios. Entre los radicales orgánicos, los nitróxidos son la clase más versátil de sondas de espín, principalmente por su tamaño relativamente pequeño, comparable a un grupo lateral de aminoácidos o nucleobase, y debido a la anisotropía hiperfina y tensor de una magnitud que es conveniente para estudiar la dinámica (Sección 10.1.4). Los radicales triarilmetilo (TAM) son químicamente incluso más inertes que los radicales nitróxido y tienen tiempos de relajación más lentos en solución líquida. Actualmente están mucho menos en uso que los radicales nitróxido, principalmente porque no están disponibles comercialmente y son mucho más difíciles de sintetizar que los radicales nitróxido.
Figura 10.1: Estructuras de sondas de nitróxido. Derivados TEMPO. Derivados PROXYL. Nitróxido de imidazolidina sensible al pH. 4 derivados de DOXYL. 5 Etiqueta de centrifugado de metanotiosulfonato (MTSL)
18.1.2. Radicales Nitróxido
El radical nitróxido se define por el grupo, que es isoelectrónico con el grupo carbonilo y así puede ser reemplazado en cálculos aproximados de campo de fuerza y dinámica molecular por un grupo. El electrón desapareado se distribuye sobre ambos átomos, lo que contribuye a la estabilidad radical, con una ligera preferencia por el átomo de oxígeno. Los radicales nitróxido se vuelven estables en la escala de tiempo de meses o años si ambas posiciones están protegidas estéricamente, por ejemplo al unir dos grupos metilo a cada uno de los átomos (Fig. 10.1). Los nitróxidos de este tipo son térmicamente estables hasta temperaturas de aproximadamente, pero se reducen fácilmente a las hidroxilaminas correspondientes, por ejemplo por el ácido ascórbico, y son inestables a pH muy bajo y muy alto. Los nitróxidos con anillos de cinco miembros (estructuras, y) tienden a ser químicamente más estables que aquellos con anillos de seis miembros (6). Los anillos de cinco miembros también tienen menos libertad conformacional que los anillos de seis miembros.
Las sondas de espín pueden dirigirse a ciertos entornos en sistemas heterogéneos mediante la elección de sustituyentes apropiados (Fig. 10.1). Las especies no sustituidas son hidrofóbicas y se distribuyen preferiblemente en ambientes no polares. La preferencia por los aceptores de enlaces de hidrógeno se logra mediante derivados de hidroxilo, mientras que los entornos iónicos pueden ser abordados por un grupo carboxilato a suficientemente alto o por un grupo trimetilamonio. Se utilizan grupos reactivos para SDSL, como el grupo metanotiosulfonato en el derivado dehidro-proxilo MTSL 5, el cual reacciona selectivamente con grupos tiol en condiciones suaves. Los grupos tiol se pueden introducir en las proteínas mediante mutación puntual dirigida al sitio de un aminoácido a cisteína y a ARN mediante el reemplazo de una nucleobase por tiouridina. En los derivados de DOXYL, un anillo de seis miembros está espiro-unido a una cadena alquílica, que puede ser parte del ácido esteárico o de moléculas lipídicas. El grupo en los derivados de DOXYL está unido rígidamente a la cadena alquílica y casi paralelo al eje de una hipotética cadena todo-trans.
18.1.3. El espectro de EPR de Nitróxido
A una buena aproximación, el sistema de espín de un radical nitróxido puede considerarse como un espín electrónico acoplado al espín nuclear del átomo del grupo. Hiperfina
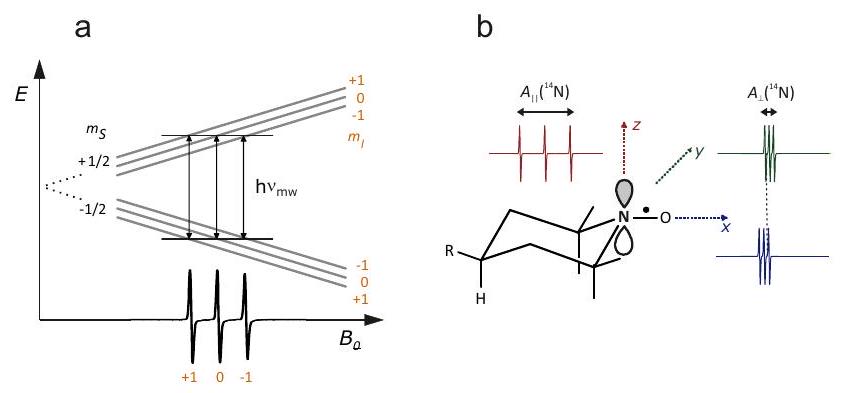
Figura 10.2: El espectro de EPR y el marco molecular de los radicales nitróxido. (a) Los subniveles hiperfinos correspondientes a los tres posibles números cuánticos magnéticos son desplazados por. Las transiciones permitidas son aquellas con y. El cuántico de microondas tiene energía constante, ya que la frecuencia de microondas es constante. Durante un barrido de campo magnético, se observa resonancia cuando la energía coincide con la diferencia de energía de los niveles de una transición permitida. Las tres transiciones corresponden a los tres posibles números cuánticos magnéticos. (b) En un sólido, cada orientación da un espectro de tres líneas, pero la división y el campo central dependen de la orientación, ya que y son anisotrópicos. A una buena aproximación, el tensor hiperfino tiene simetría axial con el eje único correspondiente a la dirección de los lóbulos orbitales en el átomo. El tensor es ortorrómbico, es decir, los espectros en el plano del marco molecular, que todos tienen la misma división hiperfina, tienen diferentes campos centrales. La dirección del enlace, que corresponde al valor máximo, es el eje del marco molecular.
los acoplamientos a otros núcleos, como los protones de metilo, no suelen resolverse y contribuyen únicamente al ensanchamiento de la línea. El acoplamiento hiperfino al átomo hibridado tiene una importante contribución isotrópica de contacto Fermi por la densidad de espín en la órbita y una contribución anisotrópica significativa de la densidad de espín en la órbita que se combina con un orbital en el átomo de oxígeno para dar al enlace N-O carácter de doble enlace parcial. La dirección de los lóbulos de la órbita se elige como eje molecular (Fig. 10.2 (b)). El tensor hiperfino tiene simetría casi axial con ser el eje único. El acoplamiento hiperfino es mucho más grande a lo largo (del orden de) que en el plano (en el orden de).
El acoplamiento espín-órbita, que induce anisotropía, surge principalmente en el átomo, donde un nivel de energía de par solitario está muy cerca del SOMO. El tensor es ortorrómbico con asimetría casi máxima. El mayor desplazamiento es positivo y se observa a lo largo del enlace N-O, que es el eje del marco molecular. Se observa un desplazamiento intermedio a lo largo del eje, mientras que el valor es muy cercano a. A frecuencias de banda X, donde la anisotropía corresponde solo a dispersión en campos de resonancia, mientras que la anisotropía hiperfina corresponde a dispersión. A frecuencias de banda W, donde, la anisotropía hiperfina sigue siendo la misma pero la anisotropía aporta una dispersión diez veces mayor de, que ahora domina.
El espectro CW EPR barrido por campo para una sola orientación se puede entender considerando la regla de selección de que el número cuántico magnético del espín electrónico debe cambiar en 1, mientras que el número cuántico magnético del espín nuclear no debe cambiar. Por lo tanto, cada transición puede asignarse a un valor de. Porque hay tres valores de este tipo,, y 1 (Fig.). La frecuencia de microondas es fija y la resonancia se observa en campos donde la energía del cuántico de microondas coincide con la energía de una transición.
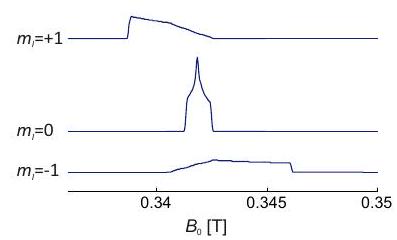
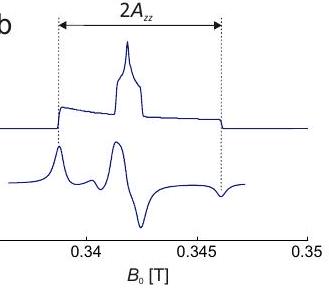
Figura 10.3: Construcción del espectro EPR en estado sólido de un nitróxido en la banda X. a) El espectro de absorción de cada transición se considera por separado. Porque, el aporte hiperfino desaparece y solo contribuye la anisotropía. Esta línea es la más estrecha en la banda. Para la dispersión por anisotropía resta de la dispersión mayor por anisotropía hiperfina. Esta línea tiene un ancho intermedio. Para la dispersión de la anisotropía se suma a la dispersión de la anisotropía hiperfina. Esta línea tiene el mayor ancho. b) Las tres contribuciones de los valores individuales se suman al espectro de absorción total de EPR (top). En CW EPR se observa la derivada de este espectro de absorción (abajo). Debido a que domina la anisotropía hiperfina, la separación entre las extremidades externas es.
Para construir el espectro de estado sólido, se debe considerar la dependencia de orientación de las tres transiciones (Fig. 10.3 (a)). En cada orientación individual, la línea es la línea central. Dado que las contribuciones hiperfinas escalan con, desaparece para esta línea y solo se observa anisotropía. En la banda X, donde la anisotropía hiperfina domina con diferencia, esta línea es la más estrecha. El lineshape es el de anisotropía pura (ver Fig. 3.4). Porque, la orientación con el mayor desplazamiento del campo de resonancia coincide con la del desplazamiento hiperfino más pequeño. De ahí que la dispersión de campo de resonancia menor por anisotropía resta de la dispersión mayor por anisotropía hiperfina. Porque, la situación es opuesta y las dos dispersiones se suman. De ahí que la transición, que en cualquier orientación dada es la línea de campo alto, tiene la mayor dispersión de resonancia, mientras que la transición de campo bajo tiene dispersión de campo de resonancia intermedia. La característica central en el espectro de absorción total (Fig.) está fuertemente dominada por la transición, mientras que los hombros externos corresponden a las transiciones (campo bajo) y (campo alto) en la orientación. Por lo tanto, la división entre las extremidades externas en el espectro CW EPR, que corresponden a estos hombros en el espectro de absorción, es.
18.1.4. Influencia de la dinámica en el espectro del nitróxido
En solución líquida, las moléculas se tambalean estocásticamente debido a la difusión rotacional browniana. A continuación consideramos la difusión rotacional isotrópica, donde la molécula se tambalea con la misma velocidad promedio alrededor de cualquier eje en su marco molecular. Esta es una buena aproximación para sondas de espín de nitroxido con sustituyentes pequeños. Por ejemplo, TEMPO con es casi esférico con un radio Van-der-Waals de ÅÅ. En agua a temperatura ambiente, el tiempo de correlación rotacional para TEMPO es del orden de. El producto con la anisotropía máxima del espectro de nitróxido EPR en un eje de frecuencia angular es mucho menor que la unidad. En esta situación, se esperan promedios de anisotropía y tres líneas estrechas de igual anchura e intensidad. El espectro en la Fig. 10.2 (a) corresponde a esta situación y una mirada más cercana revela que la línea de campo alto tiene una amplitud algo menor. Esto se puede rastrear hasta un ancho de línea mayor que para las otras dos líneas, lo que indica una más corta para la transición que para las otras transiciones. De hecho, la relajación transversal está dominada por el efecto de la combinación hiperfina y anisotropía, que es mayor para la transición que tiene la mayor dispersión anisotrópica de frecuencias de resonancia. Con el aumento del tiempo de correlación rotacional, se espera que este proceso de relajación se vuelva más fuerte, lo que debería conducir a un mayor ensanchamiento de línea que sea más fuerte para la línea de campo alto y más débil para la línea central. Esto se observa efectivamente en la simulación para ns mostrada en la traza inferior de la Fig.
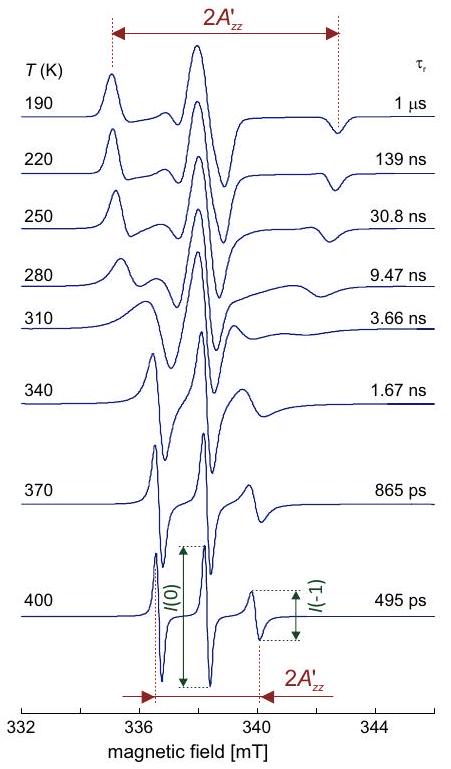
Figura 10.4: Simulación de espectros CW EPR de banda X de un radical nitróxido de volteo isotrópicamente para diferentes tiempos de correlación rotacional. Se asumió un tiempo de correlación rotacional de s at y un proceso activado con energía de activación de, cercano a los parámetros observados para TEMPO en un polímero sintético.
Según la teoría de relajación de Kivelson, la relación entre el ancho de línea de una de las líneas exteriores y el ancho de línea de la línea central viene dada por
donde
y
con el parámetro de anisotropía hiperfina
y el parámetro de anisotropía de Zeeman del electrón
El tiempo de relajación para la línea central se puede calcular a partir del ancho de línea pico a pico correspondiente en el dominio de campo como
Así, las ecuaciones (10.1-10.3) pueden resolverse para el único desconocido restante. En la práctica, las relaciones de amplitudes de línea pico a pico se analizan en lugar de las relaciones de ancho de línea, ya que se pueden medir con mayor precisión. La relación de ancho de línea está relacionada con la relación de amplitud (ver traza inferior en la Fig. 10.4) en un espectro de primera derivada por
ya que la intensidad integral de la línea de absorción (doble integral de la forma de línea derivada) es la misma para cada una de las tres transiciones. Por lo tanto, el tiempo de correlación rotacional puede determinarse, p.
donde es el ancho de línea pico a pico de la línea central. Esta ecuación se puede aplicar en el régimen de volteo rápido, donde las tres líneas individuales para, y aún pueden ser claramente reconocidas y tienen la forma de líneas de absorción derivadas simétricas.
Para un volteo más lento con, la forma de la línea se vuelve más compleja y se acerca al límite rígido (espectro de estado sólido) aproximadamente (Fig. 10.4). Estas formas lineales se pueden simular considerando el intercambio multisitio entre diferentes orientaciones de la molécula con respecto al campo magnético. A diferencia del intercambio de dos sitios, que se discute en la parte de RMN del curso de conferencias (ver Sección 3 de las notas de la conferencia de RMN), no se pueden obtener expresiones cerradas para el intercambio multisitio. Sin embargo, podemos estimar la escala de tiempo donde las características espectrales son más amplias y los tiempos de relajación transversal son los más cortos. La coalescencia para el intercambio de dos sitios se observa en. Al sustituir por la anisotropía máxima de, correspondiente a, encontramos un “tiempo de coalescencia” ns. Las simulaciones de la Fig. muestran efectivamente que alrededor de este tiempo de correlación rotacional, el
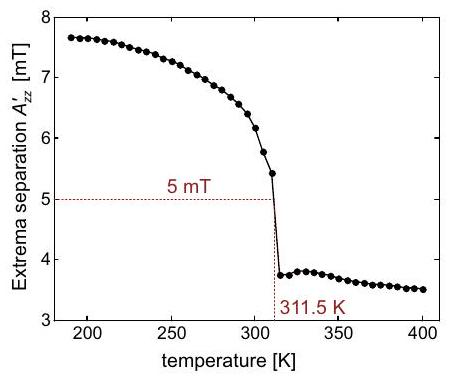
Figura 10.5: Gráfica de la separación extrema externa en función de la temperatura para espectros de nitróxido simulados bajo los mismos supuestos que en la Fig. 10.4.
el carácter del espectro cambia de intercambio rápido de orientación (espectro de tipo líquido con tres picos distintos) a intercambio de orientación lento (espectro de tipo sólido).
Una forma sencilla de analizar una dependencia de la temperatura, como la que se muestra en la Fig. 10.4, es graficar la separación extrema externa en función de la temperatura (Fig. 10.5). El “tiempo de coalescencia” en dicha parcela corresponde al gradiente más grande, el cual coincide con la media entre los valores en el límite de volteo rápido y el límite rígido, que es. En el caso que nos ocupa, este tiempo de coalescencia es y se observa a una temperatura. La temperatura es la temperatura donde el material se vuelve “blando” y las conformaciones moleculares pueden reorganizarse. Los espectros de nitróxido en el régimen de volteo lento pueden revelar más detalles sobre la dinámica, por ejemplo, si hay ejes de rotación preferidos, si el movimiento está restringido debido a la unión covalente del nitróxido a una molécula grande, o si hay orden local, como en una bicapa lipídica.
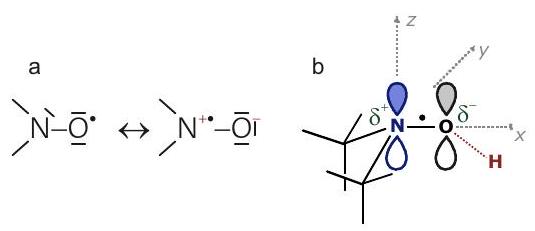
Figura 10.6: Influencia de la polaridad del ambiente y de los enlaces de hidrógeno sobre el desplazamiento y el acoplamiento hiperfino. (a) En la estructura mesomérica donde el electrón desapareado se encuentra en el átomo de oxígeno (izquierda), se asignan formalmente a cinco electrones de valencia y seis a, lo que corresponde a la electroneutralidad. En la estructura mesomérica donde el electrón desapareado se encuentra en el átomo de nitrógeno (derecha), solo se asignan formalmente cuatro electrones de valencia y siete a, lo que corresponde a una carga positiva en y a una carga negativa at. b) La mezcla de la estructura mesomérica de carga separada genera cargas parciales y se ve favorecida en un ambiente polar que criba la atracción de Coulomb de las dos cargas. El enlace de hidrógeno al oxígeno disminuye la energía del par solitario, haciendo que la excitación de un electrón de par solitario al SOMO sea menos probable y, por lo tanto, disminuye el desplazamiento.
18.1.5. Polaridad y proticidad
La deslocalización del electrón desapareado en el grupo se puede entender considerando estructuras mesoméricas (Fig. 10.6). Si el electrón desapareado reside en el oxígeno, el número formal de electrones de valencia es cinco en nitrógeno y seis en oxígeno, correspondiente a la carga nuclear que no es compensada por electrones de capa interna. De ahí que ambos átomos sean formalmente neutros en esta estructura limitante. Si, por otro lado, el electrón desapareado reside en el átomo de nitrógeno, solo se asignan cuatro electrones de valencia a este átomo, mientras que siete electrones de valencia se asignan al átomo de oxígeno. Esto corresponde a la separación de carga con la carga positiva formal sobre nitrógeno y la carga negativa formal sobre oxígeno. La forma de carga separada se ve favorecida en los disolventes polares, los cuales filtran la atracción de Coulomb entre las dos cargas, mientras que la forma neutra se ve favorecida en los disolventes no polares. Por lo tanto, para un radical nitróxido dado en una serie de disolventes, se espera que el acoplamiento hiperfino, que proviene de la densidad de espín en el átomo de nitrógeno, aumente con el aumento de la polaridad del disolvente. Este efecto efectivamente se ha encontrado. Se ve más fácilmente en el estado sólido para pero también se puede discernir en el estado líquido para.
Se espera que el cambio en esté anti-correlacionado con el desplazamiento, debido a que este desplazamiento surge del SOC en el átomo de oxígeno y, cuanto mayor es la densidad de espín en el átomo de nitrógeno, menor es en el átomo de oxígeno. Este efecto también se ha encontrado y es más fácilmente detectado por EPR de campo alto/alta frecuencia a frecuencias de frecuencias de banda W de o incluso frecuencias más altas. La forma en que se correlaciona depende de la proticidad del solvente. Los solventes próticos forman enlaces de hidrógeno con los pares solitarios en el átomo de oxígeno del grupo N-O. Esto disminuye la energía de los orbitales de pares solitarios, haciendo menos probable la excitación de un electrón de estos orbitales al SOMO. Dado que esta excitación proporciona la principal contribución al SOC y, por lo tanto, al desplazamiento, el enlace de hidrógeno al oxígeno reduce el desplazamiento. Si dos nitróxidos tienen el mismo acoplamiento hiperfino en un ambiente aprótico y prótico, será menor en el ambiente prótico. Este efecto también se ha encontrado. En algunos casos fue posible discernir etiquetas de nitroóxido con cero, uno y dos enlaces de hidrógeno por resolución de sus características en espectros CW EPR de banda W. Se han encontrado pendientes de para aprótico en ambientes próticos para la correlación entre y para MTSL en bacteriorodopsina marcada con espín en bicapas lipídicas [Ste+00].
19. Accesibilidad al agua
La polaridad y la proticidad son parámetros proxy para la accesibilidad al agua de sitios marcados por espín en proteínas. Otras dos técnicas proporcionan información complementaria. Primero, el agua puede ser reemplazada por agua deuterada y se puede medir la profundidad de modulación del deuterio ESEEM. Debido a la dependencia de la profundidad de modulación (ver Ec. (8.7)) la técnica es más sensible a los núcleos de deuterio en las proximidades de la etiqueta de espín. Siempre y cuando se sumen las contribuciones de profundidad de modulación de núcleos individuales, de modo que la profundidad de modulación de deuterio total es una medida para la concentración de deuterio local cercana a la etiqueta. Los datos se pueden procesar de una manera que elimine la contribución de los núcleos directamente unidos a hidrógeno. Estrictamente hablando, esta técnica mide la concentración no solo de protones de agua sino también de la de cualquier protón intercambiable cerca de la etiqueta, pero sólo en la medida en que estos protones intercambiables son accesibles al agua durante la preparación o medición de la muestra.
Una segunda técnica más directa que es aplicable a temperatura ambiente mide la señal de RMN protónica en función de la potencia de microondas irradiada con la frecuencia de microondas encendida resonante con la transición central de una etiqueta de giro de nitróxido. Dicha irradiación transfiere la polarización de espín electrónico a protones de agua por el efecto Overhauser. Esta polarización nuclear dinámica (DNP) de Overhauser es altamente específica para el agua, ya que depende críticamente de que la señal de RMN de protón del agua sea estrecha y de la rápida difusión del agua. En biomoléculas, la accesibilidad al agua de las etiquetas de espín es alta en las superficies expuestas al agua de proteínas solubles y de membrana y baja dentro de las proteínas y en las superficies expuestas a lípidos. Para los transportistas, la accesibilidad al agua puede cambiar con el estado en el proceso de transporte.
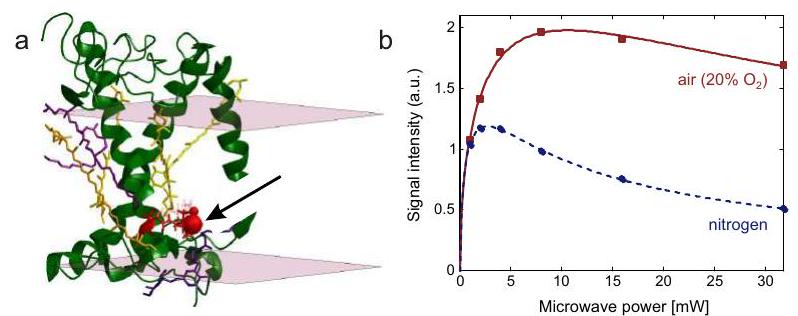
Figura 10.7: Caracterización de la accesibilidad al oxígeno en el sitio marcado por giro V229C en el complejo LHCII de cosecha de luz de planta principal por saturación progresiva de potencia CW. a) Modelo de cinta de LHCII con verde con sus cofactores carotenoides (amarillo, violeta) y modelo de relleno de espacio de residuo 229 (rojo, marcado por una flecha). Los planos rosados corresponden a la capa del grupo de cabeza lipídica de la membrana tilacoidea en cloroplastos. (b) Curvas progresivas de saturación de potencia en ausencia (azul) y presencia (roja) de oxígeno.
19.1.6. Accesibilidad al oxígeno
Dado que la colisión de oxígeno triplete paramagnético con sondas de espín mejora la relajación (Fig., el parámetro de saturación es menor para las etiquetas de espín accesibles al oxígeno que para las etiquetas de espín no accesibles al oxígeno. Este cambio puede ser detectado por mediciones progresivas de saturación de potencia CW (Sección 7.2.2). El experimento se realiza de manera más conveniente con tubos capilares hechos del plástico permeable a los gases TPX. Se realiza una medición de referencia en atmósfera de nitrógeno, lo que provoca desoxigenación de la muestra en la escala de tiempo de 15 min. Luego se cambia la corriente de gas a aire (20% de oxígeno) u oxígeno puro y se repite la medición. Dichos datos se muestran en la Fig. para el residuo 229 en el complejo principal de recolección de luz de plantas LHCII. Este residuo está expuesto a lípidos. Como molécula no polar, el oxígeno se disuelve bien en la región de la cadena alquílica de una bicapa lipídica. En consecuencia, la señal se satura a mayor potencia en una atmósfera de aire que en una atmósfera de nitrógeno. La accesibilidad al oxígeno puede cuantificarse mediante un parámetro normalizado (Sección 7.2.2).
20. Mediciones locales de pH
El acoplamiento hiperfino de las sondas de espín de nitróxido se vuelve sensible si el heterociclo que contiene el grupo también contiene un átomo de nitrógeno que puede protonarse en el intervalo deseado. Esto se aplica, por ejemplo, al nitróxido de imidazolidina 3 de la Fig., que tiene un valor de pK y exhibe un cambio en el acoplamiento isotrópico de N hiperfina entre la forma protonada (1.43 mT) y desprotonada, que puede resolverse fácilmente en solución líquida. Al modificar la sonda a un marcador, se puede medir local cerca de un residuo de interés en una proteína.
21. Trampas giradas
Muchos radicales son muy reactivos. Este hecho hace que su detección durante reacciones químicas y en células vivas sea muy importante, pero también hace que su concentración sea muy baja, ya que muchas veces su reacción de formación es más lenta que las reacciones que vuelven a destruirlas. Por ejemplo, la concentración del radical hidroxilo, una especie reactiva de oxígeno (ROS) en células vivas, es demasiado baja para la detección de EPR incluso en condiciones donde conduce a daño celular o muerte celular. La situación es algo mejor para el radical anión superóxido, pero las concentraciones fisiológicamente relevantes son difíciles de detectar también para esta especie.
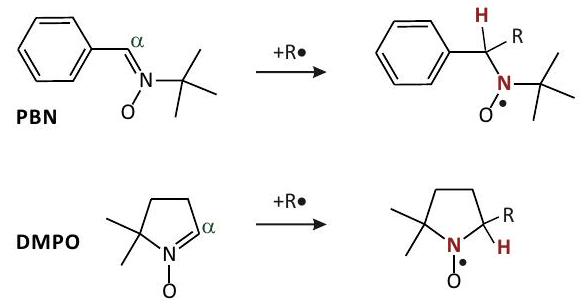
Figura 10.8: Reacción de las trampas de espín comúnmente utilizadas fenilbutilnitrona (PBN) y N-óxido de 5,5-dimetil-1pirrolina (DMPO) con radicales inestables. Los acoplamientos hiperfinos del átomo y del nitróxido formado (rojo) así como el valor del nitróxido proporcionan información de huellas dactilares sobre el tipo de radical.
Los ROS y algunos otros radicales altamente reactivos de interés se detectan más fácilmente mediante el atrapamiento de espín. Una trampa de espín (Fig. 10.8) es un compuesto diamagnético que se ceba para formar un radical estable por reacción con un radical inestable. Las trampas de espín más utilizadas son las nitronas que forman radicales nitróxido por adición del radical inestable al átomo en posición del grupo nitrona. Los radicales nitroxido formados no son tan estables como los usados como etiquetas de espín, principalmente porque contienen un átomo de hidrógeno en posición al grupo. Su vida útil suele estar en la escala de tiempo de minutos, que es suficiente para la detección. El acoplamiento hiperfino del átomo es sensible al tipo de radical primario, es decir, a la naturaleza del otro sustituyente en el átomo. Además, estas nitronas están menos abarrotadas estéricamente que las que producirían nitróxidos más estables y por lo tanto las nitronas son más reactivas y atrapan radicales con mayor facilidad. Además del acoplamiento hiperfino, el acoplamiento hiperfino del átomo del grupo es sensible a la naturaleza de. Una base de datos de resultados experimentales apoya la asignación de casos difíciles: http://tools. niehs. nih.gov/stdb/index.
Busque los “Consejos para usar la base de datos de trampas giratorias” antes de comenzar su búsqueda. El formato de palabra clave es potente, pero no muy intuitivo.
22. Bibliografía
23. Libros
[CCM16] V. Chechik, E. Carter y D. M. Murphy. Resonancia Paramagnética Electrónica. Ed. Oxford: Oxford University Press, 2016 (citada en las páginas 8, 26).
[KBE04] M. Kaupp, M. Buhl, y V. G. Malkin (Eds.) Cálculo de Parámetros de RMN y EPR: Teoría y Aplicaciones. Ed. Weinheim: Wiley-VCH, citado en la página 16).
[Rie07] Philip Rieger. Resonancia de Espín Electrónico Análisis e Interpretación. La Real Sociedad de Química, 2007, P001-173. ISBN: 978-0-85404-355-2. DOI: 10.1039/ 9781847557872. URL: http://dx. doi .org/10.1039/9781847557872 (citada en la página 37).
[WBW94] J. A. Weil, J. R. Bolton, y J. E. Wertz. Resonancia Paramagnética Electrónica. Ed. Nueva York: John Wiley & Sons, Inc., 1994 (citada en la página 8).
24. Artículos
[Cas+60] Theodore Castner y col. “Nota sobre la Resonancia Paramagnética de Hierro en Vidrio”. En: J. Chem. Phys., páginas 668-673. Dor: http://dx. doi. org/10. (citada en la página 40.
[KM85] A. K. Koh y D. J. Miller. “Constantes de acoplamiento hiperfino y parámetros atómicos para datos de resonancia paramagnética electrónica”. En: Datos atómicos y datos nucleares Tablas 33 (1985), páginas 235-253 (citada en las páginas 22,23).
[Lef67] R. Lefebvre. “Interacciones pseudo-hiperfinas en radicales”. En: Física Molecular (1967), páginas 417-426. Dor: (citada en la página 22).
[Ste+00] Heinz-Jürgen Steinhoff et al. “Estudios de EPR de campo alto de la estructura y cambios conformacionales de la bacteriorodopsina etiquetada con espín dirigida al sitio”. En: Biochim. Biofías. - Bioenergética 1457 (2000), páginas 253-262. DOI: 10. 1016/S0005 (citada en la página 82).

25. Índice
Davies ENDOR... 59
tiempo muerto
CIERVO... 68
factor de forma
DNP... 82
polarización nuclear dinámica... 82
Interacción de contacto de Fermi

g valor
electrón libre
H
aproximación de campo alto
aproximación de campo alto
ancho de línea homogéneo
La regla del perro... 37
selectividad por contraste hiperfino

Iones Kramers... 37
Teorema de Kramers

curva
energías de nivel
primer orden
régimen lineal
M

profundidad de modulación
ESEEM... 62 orbitales moleculares

selección de orientación

PELDOR... 68
saturación progresiva de potencia... 55

número cuántico
magnético
giro
26.
escaneo rápido
especies reactivas de oxígeno
parámetro de regularización... 72
27. S
curva de saturación
regla de selección
etiquetado de giro dirigido al sitio... 75
SOMO... 11
paquete de giro
acoplamiento de espín-órbita
28. T
Regularización de Tikhonov
ESEEM de dos pulsos
29.
división de campo cero.
Exportación a DOCX