
16.2: La Tercera Ley de la Termodinámica
- Page ID
- 75359
Los átomos, moléculas o iones que componen un sistema químico pueden sufrir varios tipos de movimiento molecular, incluyendo traslación, rotación y vibración (Figura\(\PageIndex{1}\)). Cuanto mayor es el movimiento molecular de un sistema, mayor es el número de microestados posibles y mayor es la entropía. Un sistema perfectamente ordenado con un solo microestado disponible tendría una entropía de cero. El único sistema que cumple con este criterio es un cristal perfecto a una temperatura de cero absoluto (0 K), en el que cada átomo, molécula o ion componente se fija en su lugar dentro de una red cristalina y no exhibe movimiento (ignorando el movimiento cuántico del punto cero).
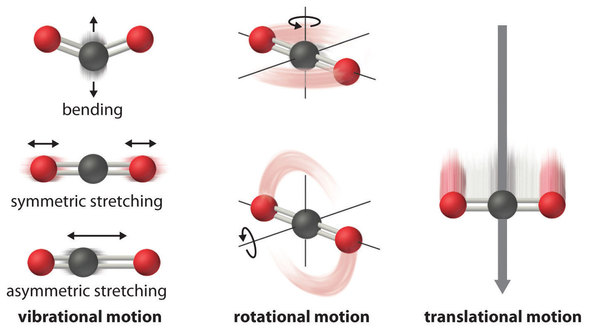
Este sistema puede ser descrito por un solo microestado, ya que su pureza, perfecta cristalinidad y completa falta de movimiento (al menos clásicamente, la mecánica cuántica argumenta por el movimiento constante) significa que no hay más que una ubicación posible para cada átomo o molécula idéntica que comprende el cristal (\(W = 1\)). Según la ecuación de Boltzmann, la entropía de este sistema es cero.
\[\begin{align*} S&=k\ln W \\[4pt] &= k\ln(1) \\[4pt] &=0 \label{\(\PageIndex{5}\)} \end{align*}\]
En la práctica, el cero absoluto es una temperatura ideal que es inalcanzable, y un monocristal perfecto también es un ideal que no se puede lograr. Sin embargo, la combinación de estos dos ideales constituye la base de la tercera ley de la termodinámica: la entropía de cualquier sustancia cristalina perfectamente ordenada en cero absoluto es cero.
La tercera ley de la termodinámica tiene dos consecuencias importantes: define el signo de la entropía de cualquier sustancia a temperaturas superiores al cero absoluto como positivo, y proporciona un punto de referencia fijo que nos permite medir la entropía absoluta de cualquier sustancia a cualquier temperatura. En esta sección, examinamos dos formas diferentes de calcular ΔS para una reacción o un cambio físico. El primero, basado en la definición de entropía absoluta proporcionada por la tercera ley de la termodinámica, utiliza valores tabulados de entropías absolutas de sustancias. El segundo, basado en el hecho de que la entropía es una función de estado, utiliza un ciclo termodinámico similar a los discutidos anteriormente.
Entropias de Estado Estándar
Una forma de calcular\(ΔS\) para una reacción es usar valores tabulados de la entropía molar estándar (\(\overline{S}^o\)), que es la entropía de 1 mol de una sustancia bajo presión estándar (1 bar). A menudo, la entropía molar estándar se da a 298 K y a menudo se demarca como\(\Delta \overline{S}^o_{298}\). Las unidades de\(\overline{S}^o\) son J/ (mol•k). A diferencia de la entalpía o la energía interna, es posible obtener valores absolutos de entropía midiendo el cambio de entropía que se produce entre el punto de referencia de 0 K (correspondiente a\(\overline{S} = 0\)) y 298 K (Tablas T1 y T2).
Como se muestra en la Tabla\(\PageIndex{1}\), para sustancias con aproximadamente la misma masa molar y número de átomos,\(\overline{S}^o\) los valores caen en el orden
\[\overline{S}^o(\text{gas}) \gg \overline{S}^o(\text{liquid}) > \overline{S}^o(\text{solid}).\]
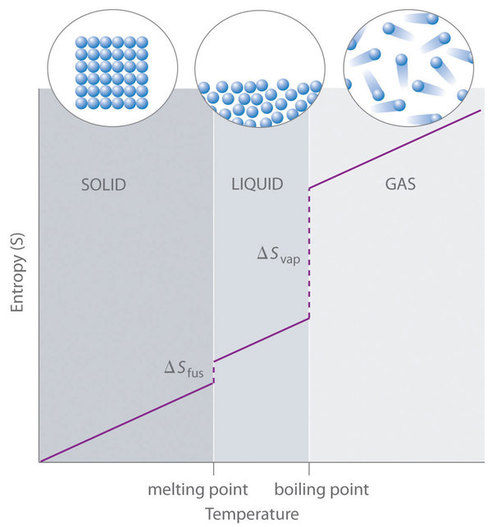
Por ejemplo,\(\overline{S}^o\) para el agua líquida es 70.0 J/ (mol•K), mientras que\(\overline{S}^o\) para el vapor de agua es 188.8 J/ (mol•K). Asimismo,\(\overline{S}^o\) es 260.7 J/ (mol•K) para los gaseosos\(\ce{I2}\) y 116.1 J/ (mol•K) para los sólidos\(\ce{I2}\). Este orden tiene sentido cualitativo basado en los tipos y extensiones de movimiento disponibles para los átomos y moléculas en las tres fases (Figura\(\PageIndex{1}\)). La correlación entre el estado físico y la entropía absoluta se ilustra en la Figura\(\PageIndex{2}\), que es una gráfica generalizada de la entropía de una sustancia frente a la temperatura.
La Tercera Ley nos permite calcular entropías absolutas
La entropía absoluta de una sustancia a cualquier temperatura superior a 0 K debe determinarse calculando los incrementos de calor\(q\) requeridos para llevar la sustancia de 0 K a la temperatura de interés, y luego sumando las proporciones\(q/T\). Se necesitan dos tipos de mediciones experimentales:
- Las entalpías asociadas a cualquier cambio de fase que pueda sufrir la sustancia dentro del rango de temperatura de interés. La fusión de un sólido y la vaporización de un líquido corresponden a incrementos importantes en el número de microestados disponibles para aceptar energía térmica, por lo que a medida que ocurren estos procesos, la energía fluirá hacia un sistema, llenando estos nuevos microestados en la medida requerida para mantener una temperatura constante (la congelación o punto de ebullición); estas entradas de energía térmica corresponden a los calores de fusión y vaporización. El aumento de entropía asociado con la transición a temperatura\(T\) es\[ \dfrac{ΔH_{fusion}}{T}.\]
- La capacidad calorífica\(C\) de una fase expresa la cantidad de calor requerida para cambiar la temperatura en una pequeña cantidad\(ΔT\), o más precisamente, en una cantidad infinitesimal\(dT\). Así, el aumento de entropía provocado por el calentamiento de una sustancia en un rango de temperaturas que no abarca una transición de fase viene dado por la suma de las cantidades\(C \frac{dT}{T}\) por cada incremento de temperatura\(dT\). Esto es, por supuesto, solo la integral
\[ S_{0 \rightarrow T} = \int _{0}^{T} \dfrac{C_p}{T} dt \label{eq20}\]
Debido a que la capacidad calorífica es en sí misma ligeramente dependiente de la temperatura, las determinaciones más precisas de entropías absolutas requieren que la dependencia funcional de\(C\) on\(T\) se use en la integral en la Ecuación\ ref {eq20}, es decir:
\[ S_{0 \rightarrow T} = \int _{0}^{T} \dfrac{C_p(T)}{T} dt. \label{eq21}\]
Cuando esto no se conoce, se puede tomar una serie de mediciones de la capacidad calorífica en incrementos estrechos de temperatura\(ΔT\) y medir el área debajo de cada sección de la curva. El área debajo de cada sección de la parcela representa el cambio de entropía asociado con el calentamiento de la sustancia a través de un intervalo\(ΔT\). A esto hay que añadir las entalpías de fusión, vaporización y de cualquier cambio de fase sólido-sólido.
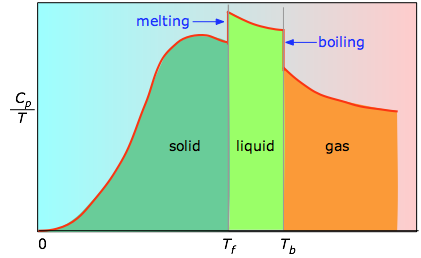
Los valores de\(C_p\) para temperaturas cercanas a cero no se miden directamente, sino que pueden estimarse a partir de la teoría cuántica. Las áreas acumulativas de 0 K a cualquier temperatura dada (Figura\(\PageIndex{3}\)) se representan luego en función de\(T\), y cualquier entropía de cambio de fase, como
\[S_{vap} = \dfrac{H_{vap}}{T_b}\]
para obtener la entropía absoluta a temperatura\(T\). Como se muestra en la Figura\(\PageIndex{2}\) anterior, la entropía de una sustancia aumenta con la temperatura, y lo hace por dos razones:
- A medida que aumenta la temperatura, se vuelven más microestados accesibles, lo que permite que la energía térmica se disperse más ampliamente. Esto se refleja en el incremento gradual de la entropía con la temperatura.
- Las moléculas de sólidos, líquidos y gases tienen cada vez más libertad para moverse, facilitando la difusión y distribución de la energía térmica. Por lo tanto, los cambios de fase van acompañados de un aumento masivo y discontinuo de la entropía.
Calculando\(\Delta S_{sys}\)
Podemos realizar mediciones calorimétricas cuidadosas para determinar la dependencia de la temperatura de la entropía de una sustancia y derivar valores absolutos de entropía bajo condiciones específicas. A las entropías molares estándar se les da la etiqueta\(\overline{S}^o_{298}\) para los valores determinados para un mol de sustancia a una presión de 1 bar y una temperatura de 298 K. El cambio de entropía estándar (\(ΔS^o\))para cualquier proceso puede calcularse a partir de las entropías molares estándar de su reactivo y especies de productos como las siguientes:
\[ΔS^o=\sum ν\overline{S}^o_{298}(\ce{products})−\sum ν\overline{S}^o_{298}(\ce{reactants}) \label{\(\PageIndex{6}\)}\]
Aquí,\(ν\) representa los coeficientes estequiométricos en la ecuación equilibrada que representa el proceso. Por ejemplo,\(ΔS^o\) para la siguiente reacción a temperatura ambiente
se calcula de la siguiente manera:
\[ΔS^o=[x\overline{S}^o_{298}(\ce{C})+y\overline{S}^o_{298}(\ce{D})]−[m\overline{S}^o_{298}(\ce{A})+n\overline{S}^o_{298}(\ce{B})] \label{\(\PageIndex{8}\)}\]
La tabla\(\PageIndex{1}\) enumera algunas entropías molares estándar a 298.15 K. Puede encontrar entropías molares estándar adicionales en las Tablas T1 y T2
| Gases | Líquidos | Sólidos | |||
|---|---|---|---|---|---|
| Sustancia | \(\overline{S}^o\)[J/ (mol•k)] | Sustancia | \(\overline{S}^o\)[J/ (mol•k)] | Sustancia | \(\overline{S}^o\)[J/ (mol•k)] |
| Él | 126.2 | H 2 O | 70.0 | C (diamante) | 2.4 |
| H 2 | 130.7 | CH 3 OH | 126.8 | C (grafito) | 5.7 |
| Ne | 146.3 | Br 2 | 152.2 | LiF | 35.7 |
| Ar | 154.8 | CH 3 CH 2 OH | 160.7 | SiO 2 (cuarzo) | 41.5 |
| Kr | 164.1 | C 6 H | 173.4 | Ca | 41.6 |
| Xe | 169.7 | CH 3 COCl | 200.8 | Na | 51.3 |
| H 2 O | 188.8 | C 6 H 12 (ciclohexano) | 204.4 | MGF 2 | 57.2 |
| N 2 | 191.6 | C 8 H 18 (isooctano) | 329.3 | K | 64.7 |
| O 2 | 205.2 | NaCl | 72.1 | ||
| CO 2 | 213.8 | KCl | 82.6 | ||
| I 2 | 260.7 | I 2 | 116.1 | ||
Un examen más detallado de Table\(\PageIndex{1}\) también revela que las sustancias con estructuras moleculares similares tienden a tener\(\overline{S}^o\) valores similares. Entre los materiales cristalinos, aquellos con las entropías más bajas tienden a ser cristales rígidos compuestos por pequeños átomos unidos por enlaces fuertes y altamente direccionales, como el diamante (\(\overline{S}^o = 2.4 \,J/(mol•K)\)). En contraste, el grafito, el alótropo más blando y menos rígido del carbono, tiene un mayor\(\overline{S}^o\) (5.7 J/ (mOL•K)) debido a más desorden (microestados) en el cristal. Las sustancias cristalinas blandas y aquellas con átomos más grandes tienden a tener entropías más altas debido al aumento del movimiento molecular y el desorden. De manera similar, la entropía absoluta de una sustancia tiende a aumentar con el aumento de la complejidad molecular debido a que el número de microestados disponibles aumenta con la complejidad molecular. Por ejemplo, compare los\(\overline{S}^o\) valores para CH 3 OH (l) y CH 3 CH 2 OH (l). Finalmente, las sustancias con fuertes enlaces de hidrógeno tienen valores menores de\(\overline{S}^o\), lo que refleja una estructura más ordenada.
La entropía aumenta con sólidos más blandos y menos rígidos, sólidos que contienen átomos más grandes y sólidos con estructuras moleculares complejas.
\(ΔS^o\)Para calcular una reacción química a partir de entropías molares estándar, utilizamos la regla familiar de “productos menos reactivos”, en la que la entropía molar absoluta de cada reactivo y producto se multiplica por su coeficiente estequiométrico en la ecuación química equilibrada. Ejemplo\(\PageIndex{1}\) ilustra este procedimiento para la combustión del hidrocarburo líquido isooctano (\(\ce{C8H18}\); 2,2,4-trimetilpentano).
Resumen
Los valores energéticos, como saben, son todos relativos, y deben definirse en una escala completamente arbitraria; no existe tal cosa como la energía absoluta de una sustancia, por lo que podemos definir arbitrariamente la entalpía o energía interna de un elemento en su forma más estable a 298 K y 1 atm de presión como cero. Lo mismo no es cierto de la entropía; dado que la entropía es una medida de la “dilución” de la energía térmica, se deduce que cuanto menor sea la energía térmica disponible para propagarse a través de un sistema (es decir, cuanto menor sea la temperatura), menor será su entropía. Es decir, a medida que la temperatura absoluta de una sustancia se acerca a cero, también lo hace su entropía. Este principio es la base de la Tercera ley de la termodinámica, que establece que la entropía de un sólido perfectamente ordenado a 0 K es cero.
En la práctica, los químicos determinan la entropía absoluta de una sustancia midiendo la capacidad calorífica molar (\(C_p\)) en función de la temperatura y luego trazando la cantidad\(C_p/T\) versus\(T\). El área bajo la curva entre 0 K y cualquier temperatura T es la entropía absoluta de la sustancia a\(T\). En contraste, otras propiedades termodinámicas, como la energía interna y la entalpía, pueden evaluarse solo en términos relativos, no en términos absolutos.
La segunda ley de la termodinámica establece que un proceso espontáneo aumenta la entropía del universo, Δ S univ > 0. Si Δ S univ < 0, el proceso es no espontáneo, y si Δ S univ = 0, el sistema está en equilibrio. La tercera ley de la termodinámica establece el cero para la entropía como el de un sólido cristalino perfecto y puro a 0 K. Con solo un microestado posible, la entropía es cero. Podemos calcular el cambio de entropía estándar para un proceso usando valores de entropía estándar para los reactivos y productos involucrados en el proceso.
Colaboradores
Paul Flowers (University of North Carolina - Pembroke), Klaus Theopold (University of Delaware) and Richard Langley (Stephen F. Austin State University) with contributing authors. Textbook content produced by OpenStax College is licensed under a Creative Commons Attribution License 4.0 license. Download for free at http://cnx.org/contents/85abf193-2bd...a7ac8df6@9.110).
Stephen Lower, Professor Emeritus (Simon Fraser U.) Chem1 Virtual Textbook