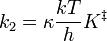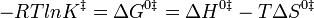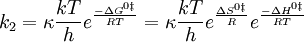5.1: Hidrógeno, Oxígeno y Agua
- Page ID
- 77475
Los fenómenos químicos deben ser tratados como si
- eran problemas en mecánica.
Lothar Meyer (1868)
Introducción
El objetivo de cada químico, sin importar con qué tipos de compuestos químicos trabaje, es comprender cómo y por qué reaccionan y cambian los químicos. Sin embargo, esta es la tarea más difícil de todas. No basta con conocer las estructuras de todos los reactivos y productos, aunque dicho conocimiento es un punto de partida vital. También debemos saber cómo las moléculas se acercan entre sí y con qué energías y con qué orientaciones interactúan. Los conceptos de energía y entropía son importantes para comprender las reacciones químicas. En este capítulo veremos algunos de los problemas que enfrentamos porque no podemos examinar eventos moleculares individuales. Examinaremos dos teorías para predecir las tasas de reacciones tan simples y comparar su éxito o falta de ellas. Observaremos los dos factores que a menudo hacen que las reacciones sean lentas -la energía y la entropía- y veremos cómo los catalizadores pueden superar estos factores y acelerar los cambios químicos. Aunque no podemos presentar una teoría completa de la reacción química (nadie puede hacer esto todavía), podemos esbozar los fundamentos sobre los que algún día se construirá esta teoría.
¿Qué sucede cuando las moléculas reaccionan?
Mecanismos de Reacción
Supongamos que podemos ver lo que sucede cuando dos moléculas reaccionan. Podemos tomar como ejemplo la reacción de una molécula de tioacetamida, CH 3 —CS—NH 2, con agua para producir acetamida, CH 3-CO—NH 2, y H 2 S (Figura 22-1).
tioacetamida, con agua para hacer acetamida, y H 2 S, (a) La molécula de tioacetamida tiene S, C y N en un plano w alrededor de una C. Los orbitales del doble enlace se distorsionan hacia S para representar su mayor elecironegatividad. Los orbitales que no juegan ningún papel en la reacción no se dibujan.]]
, con enlaces parciales de C a S y O. Los antiguos pares de electrones de enlace 0-H se están convirtiendo en pares solitarios. (c) Productos de la acetamida de reacuón y H 2 S. La geometría tetraédrica del estado de transición ha revertido a la geometría plana trigonal a medida que las hojas del átomo de S. El ángulo de enlace en H 2 S es de 92° en contraste con los 105° en (a)]] En la molécula original de tioacetamida, el átomo de carbono central está unido al carbono y al nitrógeno por enlaces σ, y por un doble enlace σ y un π al azufre (Figura 22-1a). Dado que el azufre y el nitrógeno son más electronegativos que el carbono, los electrones son más atraídos por estos átomos. Así, los pares de electrones de los enlaces carbono-azufre y carbono-nitrógeno, sus enlaces se desplazan ligeramente hacia el azufre y el nitrógeno, lo que hace que estos dos átomos porten una pequeña carga negativa y el carbono central tenga una pequeña carga positiva.
La dirección de aproximación más favorable de una molécula de agua es perpendicularmente desde ambos lados del plano de los cuatro átomos pesados. La orientación más favorable de la molécula de agua entrante es como se muestra en la Figura 22-1a. Aquí un par de electrones solitario del agua es atraído por la carga positiva sobre el carbono central. A medida que la molécula de agua se acerca a este átomo de carbono, los electrones de par solitario son atraídos hacia él y comienzan a formar un enlace parcial. Esta formación de enlaces parciales tiene dos efectos: debilita el enlace entre el carbono y el azufre al repeler los electrones aún más hacia el azufre, y al mismo tiempo debilita los enlaces O-H en el agua al tirar de los electrones de estos enlaces hacia el oxígeno a medida que los electrones del par solitario de oxígeno son atraídos hacia el carbono. Este estado intermedio aparece en la Figura 22-1b. El átomo de carbono central tiene ahora dos enlaces simples con carbono y nitrógeno, y dos enlaces parciales con azufre y oxígeno.
Este estado intermedio no es estable. Si la molécula de agua vuelve a caer y la situación vuelve a la de la Figura 22-1a (y no hay razón para que esto no pueda suceder), entonces no vemos ninguna reacción neta. La molécula de agua rebota de una colisión con tioacetamida y va por su camino separado. También podría suceder que el átomo de azufre caiga, como en la Figura 22-1c. En esta reacción, los dos protones liberados por el oxígeno ya que hace un doble enlace con el carbono son atraídos por el átomo de azufre con cuatro pares de electrones, y resulta una molécula de H 2 S. La reacción
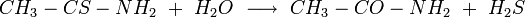
está completo. La reacción inversa también puede ocurrir; una molécula de H 2 S puede colisionar con una de acetamida y producir agua y tioacetamida. Sería menos probable que veamos tal evento si pudiéramos ver reacciones a nivel molecular, simplemente porque hay muy pocas moléculas H 2 S en comparación con el número de moléculas de agua.
Factores de Reacciones
La geometría de aproximación es un factor importante en las reacciones. Si la molécula de agua se aproximara en el plano de la molécula de tioacetamida, encontraría su entrada bloqueada por pares de electrones solitarios de azufre y átomos de hidrógeno (en mayor medida de lo que es evidente en los dibujos esqueléticos de la Figura 22-1). Además, si la molécula de agua se acercara con un átomo de hidrógeno, en lugar de un par de electrones solitario, apuntando al carbono central, no sería tan atraída por la molécula de tioacetamida y sería más probable que rebotara sin reaccionar. Si pudiéramos ver cada colisión, podríamos ver que solo 1 colisión en 10, o en 100, tenía ambas moléculas correctamente orientadas para la reacción.
Un segundo factor es la energía de las dos moléculas. En las teorías más simples esto se expresa únicamente como la velocidad relativa de las dos moléculas al colisionar. Si la velocidad relativa de las dos moléculas es pequeña al impactar, el estado intermedio será más probable que vuelva a las moléculas de partida. Una molécula de agua que se mueve lentamente puede rebotar sin daño en la tioacetamida. En contraste, una molécula de agua que choca con fuerza contra la tioacetamida tiene más posibilidades de alejar el átomo de azufre, produciendo así acetamida y H 2 S. Podríamos encontrar que podríamos trazar una curva de la probabilidad de reacción en función de la velocidad de aproximación de los dos moléculas a lo largo de una línea que conecta sus centros.
Desafortunadamente, los tipos de observaciones que hemos estado describiendo para una reacción son un sueño inalcanzable. Debemos tratar de averiguar lo que sucede durante una reacción de una manera más indirecta. Frecuentemente lo más que podemos decir sobre un mecanismo de reacción propuesto es que no es incompatible con los datos. Siempre existe la posibilidad persistente de que algún otro mecanismo de reacción pueda explicar los mismos datos también. Un ejemplo clásico de esta ambigüedad es la reacción de H 2 y I 2. En 1893, Max Bodenstein, en Alemania, estudió la reacción:

Este fue el primer estudio cinético integral de una reacción ocurrida en fase gaseosa. Desde ese momento hasta 1967, prácticamente todos los textos y tratados cinéticos utilizaron esta reacción como el ejemplo ideal de un mecanismo de colisión de dos cuerpos. Una molécula de gas de H 2 colisiona con una molécula de I 2, reorganizan átomos, y dos moléculas de HI son el resultado. Pero en 1967, J. H. Sullivan demostró que esta reacción no se da por una colisión de dos cuerpos en absoluto, sino por una complicada reacción en cadena. Veremos más adelante por qué los datos medidos antes de 1967 podrían explicarse con igual facilidad con el modelo de dos cuerpos o uno de tres cuerpos.
No solo somos incapaces de observar moléculas individuales, no podemos elegir la orientación de las moléculas en caso de colisión. Lo mejor que podemos hacer es estimar la probabilidad de que las moléculas estén adecuadamente orientadas y luego modificar nuestros cálculos de velocidades de reacción por un factor adecuado. Tal corrección a veces se usa y se llama factor estérico.
Descubriendo el proceso de reacciones y haces moleculares cruzados
En una reacción gaseosa o una reacción en solución, ni siquiera podemos elegir la velocidad de aproximación de las moléculas reaccionantes. Las moléculas en una muestra de gas tendrán una distribución de velocidades. Podemos cambiar la distribución de velocidades variando la temperatura del gas. Como se ilustra en la Figura 3-11, en gas nitrógeno la fracción de todas las moléculas que tienen una velocidad mayor que algún valor como 1000 mseg -1 aumenta a medida que aumenta la temperatura. A 273 K, solo 0.44% de las moléculas de N 2 tienen velocidades de 1000 m seg-1 o mayores; a 1273 K, 35% tiene esta velocidad o mayor; a 2273 K, esta fracción aumenta a 55%. No obstante, nada de lo que podamos hacerle al sistema nos dará una velocidad específica.
Podemos eliminar la distribución de velocidad para ciertas reacciones mediante el método de haces moleculares cruzados (Figura 22-2). En lugar de reacciones que ocurren entre moléculas dispersas en una solución o un gas, se hacen pasar haces de moléculas o iones entre sí en una cámara evacuada con cantidades insignificantes de otras moléculas presentes. Un par de ruedas con aberturas espaciadas controlan la velocidad de las moléculas. Las fuentes de haz suelen ser hornos que emiten una corriente de moléculas de gas, y campos electrostáticos que aceleran iones]] Las moléculas en los haces cruzados reaccionan entre sí y se dispersan de los haces. Los productos de la reacción, y las moléculas iniciales sin reaccionar, se pueden observar como una función del ángulo de dispersión mediante el uso de un detector móvil montado dentro de la cámara. Esta disposición tiene la gran ventaja de que los selectores de velocidad pueden limitar el haz a moléculas con velocidades en un rango pequeño elegido. El conocimiento de los productos de la reacción en función del ángulo de desviación o dispersión proporciona mucha más información sobre el proceso de reacción. El problema de orientación permanece en un experimento de haz molecular, pero uno puede imaginar experimentos en los que también se controla este factor. Los campos magnéticos o eléctricos intensos colocados justo antes de que los haces se crucen podrían dar a la mayoría de las moléculas del haz una orientación preferida en el espacio si las moléculas tienen momentos magnéticos o momentos dipolares.
Algunas de las reacciones que se han estudiado con haces moleculares cruzados son


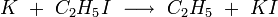
Los reactivos, haces de átomos de K, HBr, CH 3 I y moléculas C 2 H 5 I, se emiten desde hornos calentados dentro de la cámara evacuada. El detector es un filamento de alambre calentado llamado detector de ionización de superficie, que es sensible a metales alcalinos o compuestos de metales alcalinos.
Yuan T. Lee, ganador del Premio Nobel de Química de 1986, ha extendido los experimentos de haz molecular para incluir moléculas más grandes y complejas. Estudió diversas reacciones entre moléculas orgánicas y átomos de flúor u oxígeno. Su trabajo más reciente se ha centrado en las reacciones básicas relacionadas con el agotamiento del ozono. Yuan Lee ha utilizado haces moleculares cruzados para explorar más a fondo el agotamiento del ozono mediante el envío de un haz de helio sembrado con ozono (O 3) a través de un rayo láser pulsado. El rayo láser da lugar a luz UV de alta potencia y promueve la disociación de O 3 en un O neutro y O 2. Esta investigación sugiere que potencialmente podríamos controlar esta secuencia de reacciones para crear O 3 estratosférico que luego sería liberado al ambiente, reparando así la capa de ozono de la Tierra. [1]
El grupo de Lee en el Laboratorio Lawrence Berkeley también está utilizando haces moleculares para investigar procesos fotoquímicos. Los investigadores utilizan un rayo láser para excitar moléculas o átomos después de que hayan sido acelerados pero antes de que choquen, dándoles más control sobre el tipo de reacción química que se produce. También están estudiando el uso de la excitación láser durante experimentos de haz molecular para promover la eliminación de uno o más átomos específicos de moléculas más grandes así como la combustión de hidrocarburos. [2]
La investigación de haces moleculares cruzados ha facilitado una mayor comprensión de qué intermedios (las moléculas de corta duración que se forman primero en una colisión molecular pero que pronto se desintegran en una molécula más estable) se forman en una reacción y cómo. Al variar las velocidades y ángulos de los reactivos entrantes, y al medir y determinar las distribuciones de los productos resultantes, los investigadores pudieron inferir las condiciones necesarias y suficientes bajo las cuales se llevaría a cabo la reacción. Además, el haz transversal molecular ha proporcionado información sobre cómo interactúan los catálisis en las reacciones químicas. A medida que se realizan más investigaciones utilizando experimentos de haz molecular cruzado, la capacidad de controlar los productos de las reacciones químicas se está convirtiendo en una realidad. [3]
La desventaja de los experimentos con haces moleculares es que no todas las reacciones químicas son adecuadas para el estudio con haces moleculares en cámaras evacuadas. Los métodos de haz molecular siguen siendo una herramienta especial para realizar estudios completos de ciertas reacciones especiales. La mayoría de las reacciones químicas deben ser estudiadas por métodos a granel, en mezclas de gases, soluciones y (menos frecuentemente) sólidos.
Medición de las velocidades de reacción
La velocidad de reacción suele seguirse en métodos a granel observando la desaparición de un reactivo o la aparición de un producto en un tiempo dado. Si la reacción química es
- A + 2 B → 3 C
entonces la tasa de aparición del producto C en un intervalo de tiempo Δt es
![rate = \frac{\Delta [C]}{\Delta t}\,](https://upload.wikimedia.org/math/e/5/e/e5e6c0f71a1d8ce6a13b77278fbbe6c8.png) (22-1)
(22-1)
en el que la concentración de C, [C], suele expresarse en moles litro -1. Esta es la tasa promedio de aparición de C durante el intervalo de tiempo Δt. El límite de esta tasa promedio a medida que el intervalo de tiempo se hace más pequeño se llama la tasa de aparición de C en el tiempo t. Es la pendiente de la curva de [C] versus t en el tiempo t. Esta pendiente o tasa instantánea está escrita
Dado que una molécula de A desaparece por cada tres de C que se producen, y dos moléculas de B desaparecen durante el mismo proceso, las tasas de desaparición y aparición de especies químicas están relacionadas por la expresión
La velocidad de una reacción química dependerá de las concentraciones de los reactivos, aunque no siempre de la manera que podría esperarse de la ecuación química general. Para la reacción del gas hidrógeno con yodo gaseoso para producir HI,
- H 2 + I 2 → 2 HI
la relación entre tarifas es
y como se podría esperar intuitivamente, la ecuación de tasa es
![\frac{d[HI]}{dt}\,=k[H_2][I_2]](https://upload.wikimedia.org/math/d/c/6/dc691b96d33e195cb3e0a65f37cc6627.png) (22-2)
(22-2)
La velocidad de reacción es proporcional a las concentraciones de H 2 e I 2, y depende de la primera potencia de cada concentración. Esto no quiere decir que la reacción proceda por una colisión de una molécula H 2 con una I 2; desde 1967 hemos tenido pruebas de que no lo hace. Debemos distinguir claramente entre el orden de una reacción y la molecularidad de la reacción. La constante de proporcionalidad en la ecuación 22-2 se denomina constante de velocidad.
El orden de una reacción es la suma de todos los exponentes de los términos de concentración en la ecuación de velocidad. La reacción de HI es de primer orden en cada una de las concentraciones de reactivos y es de segundo orden global. El orden es un parámetro puramente experimental y describe lo que se observa sobre la ecuación de velocidad en lugar de implicar nada sobre el mecanismo de reacción.
La molecularidad de una simple reacción de un solo paso es el número de moléculas individuales que interactúan en la reacción. La molecularidad requiere un conocimiento del mecanismo de reacción. Una reacción como la del hidrógeno y el yodo en realidad puede tener lugar como una serie de media docena de reacciones individuales para las cuales podríamos especificar la molecularidad de cada una. El concepto de la molecularidad de una reacción global que ocurre en una serie de pasos no tiene sentido. La mayoría de las reacciones simples de un solo paso son unimoleculares (decaimiento espontáneo) o bimoleculares (colisión). Las reacciones trimoleculares verdaderas son raras, ya que las colisiones de tres cuerpos son poco probables. Las reacciones tetramoleculares y superiores son prácticamente inauditas. Las reacciones que parecen ser trimoleculares o superiores (a partir de su estequiometría), luego de un cuidadoso estudio, suelen ser la suma de una serie de pasos simples unimoleculares y bimoleculares. Uno de los retos de la cinética química es determinar el verdadero conjunto de reacciones en tal caso.
La reacción del gas hidrógeno con bromo contrasta completamente con la del yodo. La reacción general es similar:
- H 2 + Br 2 → 2 HBr
pero la dependencia experimental de la tasa de producción de HBr sobre las concentraciones de reactivos y productos es completamente diferente de la ecuación 22-2:
![\frac{d[HBr]}{dt}\,=\frac{k[H_2][Br_2]^{1/2}}{1+k'([HBr]/[Br_2])}\,](https://upload.wikimedia.org/math/4/0/7/40755e38469dd2283afab410434c936f.png) (22-3)
(22-3)
Esta expresión tiene dos constantes de velocidad experimentales, k y k'. No podemos hablar de la molecularidad de la reacción, porque el proceso global es el resultado de una elaborada cadena de reacciones a la que volveremos más adelante. Hasta el orden es un rompecabezas. Al inicio de una reacción de H 2 con Br 2, cuando hay poco HBr presente, se puede descuidar el segundo término en el denominador. Entonces la reacción es efectivamente 1 1/2 orden: primer orden en H 2 y medio orden en Br 2. A medida que el producto, HBr, se acumula, ralentiza la tasa de producción de más HBr. Por lo tanto, HBr se denomina inhibidor de la reacción.
La formación de HCl es aún más complicada. La producción de HCl es acelerada por luz de intensidad I y es inhibida por la presencia de gas oxígeno, incluso a bajas concentraciones de oxígeno. Durante años la dificultad de purificar los gases H 2 y Cl 2 y eliminar todas las trazas de O 2 condujo a conclusiones erróneas sobre la cinética de esta reacción. La mejor ecuación de velocidad experimental para la aparición de HCl es
![\frac{d[HCl]}{dt}\,=\frac{k_1I[H_2][Cl_2]}{k_2[Cl_2]+[O_2]([H_2]+k_3[Cl_2])}\,](https://upload.wikimedia.org/math/e/7/b/e7b026d78c8c9f65c8a4408e7d2f1ad1.png) (22-4)
(22-4)
¡Observe que, en el límite de la ausencia total de gas oxígeno, la tasa es proporcional a la concentración de gas H 2 y no depende en absoluto de la concentración de gas Cl 2! (El segundo término en el denominador de la ecuación 22-4 es cero, y las concentraciones restantes de Cl 2 en el numerador y denominador cancelan.) La reacción se complica por reacciones secundarias que tienen lugar en las superficies de los recipientes de reacción. Los resultados obtenidos a veces dependen del tamaño y forma del recipiente de reacción. Todo esto está muy lejos de la simplicidad del sistema HI. También hay reacciones secundarias en el sistema HI, pero no son importantes por debajo de los 800 K.
Siguiendo el curso de una reacción
¿Cómo medimos las concentraciones de reactivos y productos durante una reacción para encontrar ecuaciones de velocidad como las que hemos estado examinando? Si el número total de moles de gas cambia durante una reacción gaseosa, el curso de la reacción se puede medir de dos maneras: (1) el cambio de presión a volumen constante o (2) el cambio de volumen a presión constante. Estos son ejemplos de mediciones físicas que se pueden realizar en el sistema mientras éste está reaccionando. Tienen la ventaja de no molestar al sistema de reacción, y suelen ser rápidos. Con los dispositivos de grabación automática, podemos monitorear una cantidad física continuamente durante la reacción.
Otras mediciones físicas a menudo utilizadas en estudios cinéticos incluyen métodos ópticos como la rotación de la luz por una solución (útil si los reactivos y productos tienen diferentes capacidades para rotar la luz polarizada), cambios en el índice de refracción de una solución, color y espectros de absorción. Los métodos eléctricos comunes incluyen la conductividad eléctrica de una solución (especialmente útil cuando se producen o consumen iones), el potencial eléctrico en una celda y la espectrometría de masas. También se han utilizado conductividad térmica, viscosidad de una solución de polimerización, calores de reacción y puntos de congelación. La desventaja de todos estos métodos es que son indirectos. La propiedad observada debe ser calibrada en términos de concentraciones de reactivos y productos. La calibración está sujeta a errores sistemáticos, especialmente si se producen reacciones secundarias.
Se utilizan varios métodos para preparar un sistema de observación en un experimento cinético. Uno de los métodos comunes de hoy en día, llamado el método de 'salto de temperatura' o 'salto en T', fue desarrollado en la década de 1950 por el físico alemán Manfred Eigen, quien más tarde compartió el Premio Nobel de Química de 1967 por su trabajo. En el método T-jump, se contiene una pequeña muestra del sistema que se estudia y se deja reaccionar al equilibrio. Entonces la temperatura del sistema se eleva varios grados en tan solo 100 nanosegundos y la reacción se observa a medida que se ajusta a su nuevo punto de equilibrio. 100 nanosegundos resultan ser demasiado lentos para algunas reacciones, pero el método de salto en T es adecuado para una gran cantidad de procesos químicos. Los hombres con los que Eigen compartió su premio, R.G.W. Norrish y George Porter, desarrollaron el método flash-fotólisis, que era similar al método del salto en T en cuanto que proporcionaba una forma de hacer observaciones de reacciones de alta velocidad. A menudo los científicos que excitan reacciones usando el método T-jump observan mediante espectrometría; un estudio de 2009 realizado por Michael Frunzi, Hai Xu, R. James Cross y Martin Saunders utilizaron RMN para determinar la cinética de la reacción de C 60 con 9,10-dimetilantraceno.
Los métodos químicos son más directos y producen concentraciones directamente. Con tales métodos, se extrae una pequeña muestra de la mezcla reaccionante, y la reacción se detiene por dilución o enfriamiento de la mezcla el tiempo suficiente para medir las concentraciones. Una seria desventaja es que estamos quitando una parte del sistema de reacción y con ello cambiándolo gradualmente. Además, si la reacción no se puede detener en la muestra que se retira para su análisis, entonces el análisis es mucho menos preciso. En las reacciones en fase gaseosa entre H 2 y Cl 2, Br 2 o I 2, no hay cambio en el número de moles de gas antes y después de la reacción, por lo que no se pueden usar métodos de cambio de presión o volumen. Para estudiar estas reacciones, se toman muestras y las mezclas de gases se analizan químicamente para determinar sus composiciones.
Una ecuación de tasa de primer orden y la decadencia de 14 C
- Artículo de Wikipedia: Carbon-14
En un proceso de primer orden, la tasa de desaparición de un reactivo es proporcional a la cantidad del reactivo presente. Cada molécula reaccionante tiene la misma probabilidad de descomposición en un intervalo de tiempo dado; la tasa total de descomposición simplemente depende de cuántas moléculas están presentes. La expresión es
siendo n el número total de moléculas presentes. Esta expresión de velocidad se puede integrar para producir la concentración en función del tiempo:
Esta ecuación de tasa se utiliza en el ejemplo de datación con 14 C en la sección 23.5, donde las expresiones en términos de la concentración de 14 C son
![\frac{d[^{14}C]}{dt}=-k[^{14}C]\,](https://upload.wikimedia.org/math/6/5/7/6574f2e924a5302490bb2aca8d003fa0.png) (22-5)
(22-5)
![[^{14}C]=[^{14}C]_0e^{-kt}\,](https://upload.wikimedia.org/math/f/4/b/f4b5fc7e6145d8b598de9c376de1feea.png) (22-6)
(22-6)
La ecuación integrada 22-6 se grafica en la figura 23-3. Tomando el logaritmo de ambos lados de la ecuación 22-6 rendimientos
![ln[^{14}C]=[^{14}C]_0-kt\,](https://upload.wikimedia.org/math/e/3/b/e3b0f538820438fd14a6eeda746f3a40.png) (22-7)
(22-7)
Esta es la ecuación de la figura 23.8. La parcela es una línea recta, con una pendiente negativa igual a la constante de velocidad, k. La pendiente de la gráfica de la ecuación 22-6 en cualquier momento t, como se muestra en la figura 23-3, es proporcional a la concentración de 14 C restante en ese momento. Esto, en palabras, es lo que significa la ecuación 22-5.
Una visión alternativa Si bien esta es la solución tradicional para la integración del proceso de primer orden, el uso de la constante de Eulers en esta situación introduce un error sistemático en las constantes cinéticas reportadas.
Como se indicó anteriormente, en un proceso de primer orden, la tasa de desaparición de un reactivo es proporcional a la cantidad del reactivo presente.
Por lo tanto, la constante cinética debe representar la fracción de la población de reactivo presente que se descompondrá en un periodo de tiempo determinado y la fracción debe ser menor a uno.
Para tasas que son muy pequeñas en comparación con la población total la ecuación tradicional funciona bastante bien.
Por ejemplo, por simplicidad, si se supone que la población inicial es 1 (n o =1) y restringimos el periodo de tiempo al primer intervalo (t=1), y examinamos una tasa de conversión del 5% por periodo de tiempo supondríamos un resto del 95% del reactivo original después del primer periodo de tiempo. Sin embargo insertando estos términos obtenemos
Para obtener el resto esperado del 95% esta ecuación requiere que la constante cinética se incremente a 5.129%.
El problema se hace más evidente cuanto mayor sea la tasa observada, por ejemplo, si la tasa fue del 95%, entonces el reactivo restante después de un periodo de tiempo se esperaría que fuera el 5% de la población inicial inicial sin embargo,
Para producir el 5% restante esperado se debe incrementar la constante de velocidad a k=2.99573 una tasa de aproximadamente 300%, lo que puede ser difícil de observar.
Los problemas introducidos por el uso de esta ecuación pueden superarse reconociendo la división artificial de la constante ya que tanto la constante eulers como la constante cinética son constantes y no cambian, por lo que una constante elevada a una constante es una constante. Esta constante representa la fracción de la población reaccionante restante (%RP) por periodo de tiempo por lo que también se puede reescribir para incorporar la tasa de la fracción de la población que se descompondrá (%BD) por periodo de tiempo también.
Por lo tanto, la ecuación cinética para la cinética de primer orden se puede reescribir como
Donde la fracción de la población que se descompondrá (%BD) puede expresarse como la velocidad de reacción observada (r) dividida por la concentración inicial de reactivo n o.
Esta notación relaciona la constante cinética directamente con la velocidad observada y reconoce que la constante cinética no puede exceder de 1 ya que la velocidad nunca puede ser un valor mayor que el número en los reactivos iniciales de partida. [4]
Descomposición de N 2 O 5
 Cuadro 22-1
Cuadro 22-1En la sección 16.5, encontramos la descomposición del sólido N 2 O 5 como ejemplo de una reacción espontánea pero fuertemente endotérmica. Ahora veamos la descomposición del N 2 O 5 disuelto en tetracloruro de carbono como ejemplo de una reacción química de primer orden. El sólido N 2 O 5 y un producto, NO 2, son solubles en CCl 4; el otro producto, O 2, no lo es. La reacción
- N 2 O 5 → 2 NO 2 + O 2 (g)
puede ser seguido midiendo el volumen total de gas oxígeno que burbujea fuera de la solución.
Los datos para esta reacción se dan en la Tabla 22-1, después de que las mediciones de volumen de O 2 se hayan convertido en concentraciones de N 2 O 5 dejadas en la solución. Estos datos se representan gráficamente en la Figura 22-3 como ejemplo de la forma en que se tratan los datos de concentración. La figura muestra la concentración de N 2 O 5 en cualquier momento, la tasa de cambio en esta concentración, y esta tasa de cambio dividida por la concentración. Esta última cantidad es igual a la constante de tasa. Que la velocidad de cambio dividida por la concentración sea constante (dentro de los límites del error experimental en los datos de la Tabla 22-1) demuestra que la reacción es efectivamente de primer orden.
Estequiometría y expresiones de velocidad
 Figura 22-3
Figura 22-3La reacción
- 2 NO (g) + O 2 (g) → 2 NO 2 (g)
tiene una ecuación de tasa observada de la forma
La reacción es de segundo orden en NO y primer orden en O 2, y es de tercer orden general. La ecuación de velocidad coincide con la estequiometría de la reacción química; este acuerdo sugiere (pero no prueba) que esta puede ser una reacción simple de un solo paso que involucra tres moléculas. En contraste, el etanol y el decaborano reaccionan en solución según la ecuación
- 30 C 2 H 5 OH + B 10 H 14 → 10 B (OC 2 H 5) 3 + 22 H 2
Uno podría esperar ingenuamente que esto tenga una expresión de tasa de trigésimo primer orden. De hecho, la reacción es de segundo orden, primer orden en cada uno de los dos reactivos. Por la tasa de desaparición del etanol,
Los objetivos de la cinética química
Algunos procesos químicos son reacciones simples de un solo paso que involucran una, dos u ocasionalmente tres moléculas. Muchos más procesos son la combinación de varias reacciones tan simples. Uno de los objetivos de la cinética química es averiguar cuál es el verdadero mecanismo molecular de un proceso complejo. ¿Por qué el HI, HBr y HCl tienen ecuaciones de velocidad experimentales tan diferentes para una reacción que se ve superficialmente igual en los tres casos? A un cinetista la pregunta: “¿Cuál es el mecanismo de la reacción?” significa esto: “¿Cuál es la secuencia de reacciones simples que produce la cinética y estequiometría observada de la reacción global?” A esta pregunta los químicos orgánicos e inorgánicos han agregado: “¿Cuál es la geometría de la reacción para cada simple paso en el proceso general?” El objetivo de esta investigación es predecir por qué las reacciones simples proceden como lo hacen y predecir las tasas a las que ocurren. Las teorías que se han desarrollado para calcular las constantes de velocidad para reacciones simples unimoleculares y bimoleculares son nuestro siguiente tema.
Cálculo de constantes de velocidad a partir de información molecular
Supongamos una simple reacción bimolecular,
con una expresión de velocidad,
¿Hasta dónde podemos llegar en el cálculo de k a partir de las propiedades moleculares de A, B, C y D? Una de las primeras observaciones fue que k varía con la temperatura; la constante de velocidad es mayor, y la velocidad de reacción es más rápida, a temperaturas más altas.
Energía de activación de Arrhenius
La energía de activación de Arrhenius por definición es la energía que debe superarse para que ocurra una reacción química. La energía de activación de una reacción generalmente se denota en unidades de kilojulios por mol. Ahora se discutirá la teoría e historia de la energía de activación de Arrhenius. Si trazamos el logaritmo de la constante de velocidad frente al recíproco de temperatura, generalmente obtenemos una línea recta. Si bien Arrhenius no fue la primera persona en hacer esto, desarrolló la idea y le dio una explicación. Por lo tanto, tal parcela se llama trama de Arrhenius. ¿Qué significa en términos de mecanismos de reacción? Van 't Hoff y otros habían estado trabajando, a finales del siglo XIX, en la variación del cambio de energía libre de la reacción y de la constante de equilibrio con la temperatura. Descubrieron que la constante de equilibrio, K eq, varía con la temperatura absoluta, T, y con el calor de reacción de la siguiente manera:
Esta expresión se puede derivar de la ecuación de Gibbs-Helmholtz y, en última instancia, puede derivarse rigurosamente de la termodinámica. Durante el mismo periodo, G. M. Guldberg y P. Waage encontraron que podían derivar la constante de equilibrio a partir de argumentos cinéticos. Si la reacción directa en la reacción bimolecular general anterior tiene la velocidad
y la reacción inversa tiene la velocidad
asumieron que el equilibrio era el estado en el que las tasas de avance y retroceso son iguales, por lo que no se está produciendo ningún cambio neto en el sistema de reacción con el tiempo:
La constante de equilibrio, en este argumento, es la relación de las constantes de velocidad para las reacciones hacia adelante y hacia atrás. Esta es una derivación errónea. Es válido sólo cuando la reacción es un proceso simple de un solo paso en el que la estequiometría de la reacción se refleja en los coeficientes de los términos de concentración en la ecuación de velocidad. Sin embargo, es válido para el tipo de reacciones que estamos considerando aquí: reacciones bimoleculares simples. Si la constante de equilibrio es la relación de constantes de velocidad hacia adelante e inversa, la ecuación anterior sugiere que la entalpía de reacción podría ser la diferencia entre dos energías, E hacia adelante y E inversa: G°=E hacia adelante —E inversa Si la energía de Arrhenius de la activación no es una función de la temperatura, esta ecuación predice que una gráfica de In k contra el recíproco de la temperatura absoluta generará una línea recta. Esto es cierto para muchas reacciones, pero no todas, y la energía de activación es uno de los parámetros experimentales estándar mediante los cuales se describe una reacción química. Una representación simple, pero precisa, de la dependencia de la temperatura de la velocidad de una reacción química se puede expresar en la siguiente fórmula.
Esta expresión se puede reorganizar para producir la ecuación de Arrhenius más común.
Una generalización históricamente útil apoyada por la ecuación de Arrhenius es que, para muchas reacciones químicas comunes a temperatura ambiente, la velocidad de reacción se duplica por cada aumento de temperatura de 10 grados Celsius. La energía de activación es una barrera que las moléculas colisionantes deben superar si van a reaccionar en lugar de retroceder unas de otras.
Utilizando la reacción del agua con tioacetamida podemos postular que si la tioacetamida y las moléculas de agua no chocan de frente con suficiente energía la redistribución de los enlaces y la reacción posterior nunca ocurrirá. El agua retrocederá de la molécula de tioacetamida y no se llevará a cabo ninguna reacción. Este retroceso se debe a que se proporcionó energía insuficiente para superar la necesaria energía de activación de Arrhenius. Con esto establecido tenemos evidencia experimental, en la forma de la dependencia de la temperatura de k, de que parte de dicha energía umbral está involucrada en reacciones químicas.
La explicación de Arrhenius de las energías de activación supone que cada par de moléculas con energía menor que Ea no reaccionará, y cada par con energía mayor que E a reaccionará. Nada cambia en la termodinámica de la reacción general. Las barreras de activación para las reacciones hacia adelante y hacia atrás, E hacia adelante y E inversa, son tales que su diferencia, ΔH° = E hacia adelante — E inversa, es el calor termodinámico de reacción. Cuanto mayor sea la barrera, E hacia adelante, más lenta será la reacción hacia adelante. Sin embargo, dado que E reverse debe subir en la misma cantidad que E hacia adelante si su diferencia es fija, la reacción inversa se ralentiza en la misma cantidad.
El punto de equilibrio se ve afectado no por los valores numéricos individuales de las energías de activación para reacciones directas e inversas, sino solo por la diferencia entre ellas, que es ΔH°.
Teoría de Colisiones de Reacciones Bimoleculares de Gas
El siguiente paso lógico es construir una teoría de colisión para las reacciones gaseosas. Una reacción entre dos moléculas ocurre, en esta teoría, cuando las moléculas chocan con energía superior a E a. Una teoría difícilmente podría ser más sencilla. Hay dos preguntas a responder antes de que se pueda calcular la constante de tasa:
1. ¿Con qué frecuencia chocan dos moléculas por centímetro cúbico de mezcla de gases?
2. ¿En qué fracción de estas colisiones la energía combinada de las dos moléculas supera E a?
La frecuencia de colisión se puede calcular a partir de la teoría cinética simple de los gases con los métodos que se introdujeron en el Capítulo 3. La frecuencia depende de las concentraciones de los dos gases que reaccionan, y también de sus pesos moleculares, la distancia entre los centros moleculares en colisión y de la raíz cuadrada de la temperatura. Dado que las moléculas se mueven más rápidamente a temperaturas más altas, chocan con más frecuencia. La fracción de pares de moléculas que tienen energía igual o mayor que E a tras la colisión es
De acuerdo con la teoría simple de la colisión, la velocidad de reacción entonces es
La velocidad de una reacción es mayor a temperaturas más altas porque las colisiones son más frecuentes y porque la probabilidad de que un par colisionante tenga una energía mayor que E también es mayor. La constante, Z, se puede calcular a partir de los pesos moleculares y los diámetros de las moléculas reaccionantes aproximándolas con esferas. La constante de velocidad bimolecular, k, entonces es
Esta teoría se prueba en los datos del Cuadro 22-2. La energía de activación de Arrhenius se tabula para seis reacciones bimoleculares de gases, junto con el factor preexponencial observado, pz, y su valor teórico calculado a partir de la teoría de colisión y la teoría de la tasa absoluta que discutiremos en las siguientes secciones.
 Cuadro 22-2
Cuadro 22-2Hay que tener en cuenta que estos son los logaritmos de Z que se tabulan, por lo que un desacuerdo entre teoría y experimento de 1.0 significa un error por un factor de 10 en la constante de velocidad. El acuerdo es generalmente alentador para una teoría tan simple que no tiene otros supuestos que los de la teoría cinética de los gases. Hay discrepancias; por ejemplo, la velocidad de reacción de ClO es incorrecta por un factor de 400. Cuando ocurren discrepancias, la teoría de la tasa absoluta generalmente hace un mejor trabajo de predicción de Z que la teoría de iones collis.
En la columna derecha del Cuadro 22-2 se encuentran las entalpías estándar o calores de reacción. La entalpía relativa de los reactivos y productos, y la barrera de acción entre ellos, se representan gráficamente para estas reacciones en la Figura 22-5. Algunas reacciones, como NO 2 + CO, deben superar una considerable barrera de iones activat. Para otras reacciones, la barrera es inexistente, como con 2Cl0. Para otros como 2NO 2, la barrera a la reacción es solo el calor de reacción en sí, y la reacción inversa tiene una energía de activación cero. El caso más general se esquematiza en la parte inferior de la Figura 22-5.
 Figura 22-5
Figura 22-5 Tabla Molecular
Tabla Molecular Figura 22-6
Figura 22-6Complejos activados
Antes de considerar la teoría de la tasa absoluta, debemos observar más de cerca el estado de las moléculas reaccionantes a medida que cruzan la barrera de activación. En la reacción
los átomos de Cl y O están unidos al principio, y las dos moléculas de ClO están demasiado separadas para ejercer alguna influencia la una sobre la otra. Al final de la reacción, los átomos de Cl están separados por 1.99 A en una molécula de Cl 2, los átomos de O están separados por 1.21 A en una molécula de O 2, y estas dos moléculas están muy separadas. ¿Cuál es el estado intermedio, activado?
El complejo activado se esquematiza en la Figura 22-6b. Los cuatro átomos están separados por una distancia no especificada, algo más alejados que si se unieran en el sentido estable del término. Podemos estar seguros de que el complejo activado no es uno en el que los cuatro átomos estén tan separados que no ejerzan influencia el uno sobre el otro; debe existir algún tipo de complejo suelto. La base de esta afirmación es un conocimiento de las energías de enlace de las tres moléculas, y que la energía de activación para la reacción 2ClO es cero. Las energías de enlace, o las energías requeridas para separar completamente los átomos en una molécula diatómica, se muestran en la tabla molecular.
Si durante la reacción las dos moléculas de ClO se separaran primero, y luego los átomos aislados se combinaran en Cl 2 y O 2, la energía de activación para esta reacción sería el doble de la energía de enlace de ClO, o 540 kJ por 2 moles de ClO. En cambio, la energía de activación es cero. El complejo activado debe ser una combinación de los cuatro átomos de tal manera que cualquier inestabilidad creada como Cl y O separados sea inmediatamente compensada por la influencia estabilizadora de las asociaciones entre Cl y Cl y entre O y O.
Podemos pensar en el complejo activado como una “molécula” inestable, con muchas de las propiedades de una molécula, excepto que se descompone espontáneamente ya sea en reactivos o en productos. Las moléculas de tioacetamida y agua en la Figura 22-lb están en un complejo activado, y la energía de este complejo es mayor que la de la tioacetamida y el agua o la de la acetamida y sulfuro de hidrógeno.
Superficies de energía potencial
La reacción de 2ClO sugiere que en principio deberíamos ser capaces de calcular la energía potencial total de una colección de átomos en función de sus posiciones en el espacio. Este cálculo produciría una superficie de energía potencial con cerros y mesetas de alta energía, y valles de baja energía. Cualquier región de un mínimo de energía potencial en esta parcela representará una molécula estable. Incluso con cuatro átomos como en la reacción 2Cl0 necesitaríamos, desafortunadamente, seis variables para describir la disposición de los átomos: las longitudes de enlace de cada átomo a los otros tres, por ejemplo. Nuestra trama de energía potencial tendría que estar en un espacio de siete dimensiones. Esto es difícil de visualizar e imposible de construir. Necesitamos un ejemplo con solo dos variables para que el mapa se pueda trazar en el espacio tridimensional. Uno de los primeros mapas a calcular, por Henry Eyring en 1935, es la superficie de energía potencial para la reacción
en el que los tres átomos están limitados a estar en línea recta. Las únicas variables son las distancias desde el átomo de hidrógeno central a los otros dos, r 1 y r 2; siendo r 1 la distancia entre los dos primeros átomos, mientras que r 2 es la distancia entre el segundo y tercer átomos.
La energía potencial del sistema de tres átomos en función de r 1 y r2 se muestra en la Figura 22-7a.
 Leyenda 22-7
Leyenda 22-7 Figura 22-7
Figura 22-7 Figura 22-8
Figura 22-8Las disposiciones reales de los tres átomos en los seis puntos numerados marcados en color se dibujan en la Figura 22-8. Las secciones a través de esta superficie de energía potencial a valores fijos de r 1 se muestran en la Figura 22-7b. Si r 1 o r 2 es grande, los tres átomos de hidrógeno existen como una molécula H 2 y un átomo iso1ado. La sección de energía potencial a la constante r 1, para r 1 mayor que 3 A en la Figura 22-7b, es la misma que para una molécula H 2 aislada en la Figura 12-2. A medida que un átomo se acerca a una molécula H 2 desde la derecha (puntos 1 y 2 de las Figuras 22-7 y 22-8), el primer efecto notable es un aumento en la energía potencial del sistema de tres átomos.
El átomo entrante es repelido por la molécula, y se produce una situación más estable si el átomo rebota y se aleja de nuevo. Si el átomo tiene suficiente energía cinética para seguir acercándose a la molécula H 2, comenzará a debilitar el enlace H-H en la molécula H2.
En el punto 3, ambos átomos externos están ligeramente más alejados del central que una longitud de enlace H — H normal, pero la energía potencial del sistema de átomos es 25 kJ /mol mayor que la del H 2 aislado y H. El punto 3 es el complejo activado para la reacción. El complejo activado puede descomponerse ya sea en productos o en reactivos. No hay razón para que los tres átomos en el estado del punto 3 no puedan regresar al punto 1 así como proceder al punto 5. Lo cierto es que el complejo activado es inestable y debe descomponerse. Los puntos 1 a 5 en la Figura 22-7 están conectados por una línea discontinua coloreada llamada vía de reacción. Si trazamos energía potencial a lo largo de esta vía, se obtiene una curva de barrera de energía de activación, como las de la Figura 22-5. Observe que el camino de reacción en todo momento es un camino a lo largo de un valle entre paredes empinadas. Puede tomar 25 kJ de energía para construir el complejo activado del punto 3, pero toma más de 400 kJ para separar los átomos como en el punto 6. Incluso con reacciones de moléculas tan complicadas que no podemos calcular o incluso trazar su superficie de energía potencial multidimensional completa, tales perfiles siguen siendo útiles.
Teoría de Tasa Absoluta
En la teoría de la tasa absoluta, también conocida como teoría del estado de transición, se produce una reacción cuando un complejo activado se descompone en productos. Por lo tanto, la velocidad de reacción es producto de tres factores:
1. La concentración de complejos activados por centímetro cúbico
2. La tasa de descomposición de los complejos individuales o su velocidad de paso sobre la barrera de energía de activación
3. La probabilidad de que una avería vuelva a formar productos y no reactivos
Dado que el complejo activado representa un estado inestable de transición entre reactivos y productos, a menudo se le llama estado de transición. La teoría del estado de transición asume un equilibrio entre los reactivos y el complejo activado, generalmente representado por una daga doble superíndice:
Por lo tanto, la concentración del complejo activado viene dada por:
La tasa de descomposición es más complicada de calcular, pero resulta ser una constante universal para todas las reacciones bimoleculares a una temperatura dada:
en la que k = la constante de Boltzmann, y h = la constante de Planck. La probabilidad de que un desglose sea a productos y no a reactivos es el coeficiente de transmisión, m también a menudo escrito como κ. Se puede estimar únicamente como teniendo un valor entre 0.5 y 1.0 en la mayoría de las reacciones. Por lo tanto, la velocidad global de reacción es
y la constante de velocidad, k 2, es
(El símbolo k 2 se usa aquí en lugar de k para representar una constante de velocidad bimolecular para evitar confusiones con la constante de Boltzmann). Es posible calcular la constante de equilibrio, K ‡, entre los reactivos y el complejo activado a partir de propiedades moleculares observando la interpretación termodinámica de esta expresión de velocidad constante. La constante de equilibrio está relacionada con la energía libre estándar de formación del complejo activado a partir de reactivos, y esto a su vez está relacionado con la entalpía y entropía estándar de la formación del complejo activado:
Así, la constante de velocidad bimolecular, k 2, puede escribirse
La entalpía de activación, ∆H o ‡, es casi la misma cantidad que la energía de activación, E a. La diferencia es irrelevante es esta discusión. La ecuación anterior indica que la velocidad de reacción es más lenta si la energía de activación es grande. Este resultado ya se obtuvo en la teoría de la colisión; si la energía de activación es grande, solo una pequeña fracción de las moléculas tendrá suficiente energía para superar la barrera y reaccionar en lugar de rebotar tras la colisión. La ecuación también sugiere que la velocidad de reacción es más rápida si la entropía de activación es grande. Si el complejo activado está mucho más desordenado que los reactivos, la reacción se potencia debido a que la constante de equilibrio para la formación del complejo es grande, y más del complejo está presente. Por el contrario, si los reactivos están severamente restringidos cuando se combinan para hacer el complejo activado, entonces la reacción se inhibe. Podemos suponer que la entropía de activación en la reacción tioacetamida-más-agua es negativa ya que las dos moléculas se combinan para formar una unidad en el complejo. Ambas moléculas están severamente limitadas en sus orientaciones iniciales si van a construir el complejo activado con éxito.
La entropía de activación en reacciones bimoleculares es casi siempre grande y negativa porque los dos reactivos pierden entropía cuando se combinan en el complejo. A menudo, la aplicación más útil de la teoría de tasa absoluta no es calcular la constante de velocidad directamente, sino usar la constante de velocidad observada y la ecuación anterior para calcular la entropía de activación. La entropía de activación proporciona información sobre la estructura del complejo activado. Por ejemplo, si la entropía calculada de activación es positiva, entonces cualquier mecanismo que conduzca a través de un complejo activado estrechamente organizado debe ser rechazado. Como ejemplo, en la siguiente sección consideraremos dos reacciones del tipo:
que proceden por diferentes mecanismos, dependiendo de la naturaleza de los grupos R. En un mecanismo, el Br- se aleja a medida que se aproxima OH-, de la misma manera que la reacción de tioacetamida. El complejo activado es entonces una combinación de R 3 C-Br y OH -. Esto se denomina reacción asociativa o S N 2, lo que significa que es una sustitución de un grupo por otro, que los grupos son nucleófilos (donando electrones y atrayendo núcleos: bases de Lewis, de hecho), y que dos moléculas están involucradas en la reacción.
El otro mecanismo es que la molécula R3C-Br se disocie espontáneamente en Br y lo que se conoce como un ion carbonio, R 3 C, y que los iones OH- reaccionen rápidamente en una etapa separada con cualquier ion carbonio libre. El complejo activado o estado de transición en este mecanismo será el reactivo R 3 C—Br justo antes de la disociación. Esto se denomina mecanismo disociativo o S N 1 ya que es una sustitución nucleofílica en la que el paso más lento implica la disociación de una sola molécula. Uno debe ser capaz de distinguir entre estos dos mecanismos por sus entropías de activación, las cuales se pueden calcular a partir de la ecuación anterior y las constantes de velocidad medidas. El mecanismo S N 2 tendrá una gran entropía negativa de activación ya que el complejo activado se forma combinando dos moléculas. Por el contrario, el mecanismo SN1 tendrá virtualmente una entropía de activación cero debido a que el complejo activado difiere solo ligeramente de la molécula reaccionante.
Control Láser de Reacciones Químicas
Dado un conjunto de moléculas que pueden combinarse de dos formas posibles, los científicos se preguntan cómo pueden usar de manera más efectiva un láser para impulsar la reacción química de una manera u otra. Debido a que la interferencia de las vías moleculares es la clave para gobernar las reacciones, cualquier escenario láser que induzca tal interferencia puede servir como un medio para controlar las reacciones. En lugar de hacer brillar dos haces constantes sobre un objetivo, uno podría usar pulsos ultracortos de luz láser. Los láseres modernos pueden generar ráfagas tan cortas como 10 -15 segundos. A diferencia de la radiación de onda continua, un pulso de luz se compone de una colección de frecuencias distintas y, por ende, de una colección de fotones con diferentes energías. Tal luz también tiene una propiedad quizás contraintuitiva. Cuanto más breve sea el pulso, más amplio será el rango de energías dentro de él.
Esta propiedad juega un papel importante en los métodos de láser pulsado para controlar los resultados de las reacciones químicas. Al entregar un rango de energías, un pulso puede inducir movimiento (como vibración o rotación) en una molécula, lo que a su vez afecta la forma en que interactúa con otros pulsos de luz. Ordinariamente una molécula existe en un valor energético específico. Un sistema en una de estas energías fijas reside en un llamado estado estacionario y no se mueve con el tiempo. Para que una molécula sufra la dinámica, debe vivir en varios niveles de energía a la vez. Tal ensamblaje de niveles de energía se llama estado de superposición. La función de onda que describe el estado de superposición es la suma de funciones de onda que representan estados estacionarios de diferentes energías. Para construirlo, los investigadores brillan un pulso de luz láser coherente sobre la molécula. La forma en que se mueve la molécula depende de la naturaleza del pulso de luz y su interacción con la molécula. Así, podemos afectar los cambios dinámicos en la molécula cambiando la contribución relativa de las frecuencias que componen el pulso, es decir, dando forma al pulso.
Diversos investigadores han desarrollado estas ideas. Entre ellos se encuentran Stuart A. Rice de la Universidad de Chicago, Robert J. Gordon de la Universidad de Illinois en Chicago, Herschel Rabitz de la Universidad de Princeton, Ronnie Kosloff de la Universidad Hebrea de Jerusalén y Kent R. Wilson de la Universidad de California en San Diego. Sus resultados muestran que los pulsos construidos a partir de una complicada mezcla de frecuencias son necesarios para controlar la dinámica molecular de manera óptima, pero las aproximaciones simples a menudo bastan para romper las moléculas de manera controlada.
Reacciones Complejas
La mayoría de las reacciones químicas no son simples reacciones unimoleculares o bimoleculares, sino combinaciones de éstas. Es por ello que surgen ecuaciones de tasa tan complicadas como las ecuaciones 22-3 o 22-4. Incluso la reacción de hidrógeno-yodo, que se ha utilizado durante más de medio siglo como ejemplo clásico de una simple reacción bimolecular (ecuación 22-2), es compleja.
La reacción de hidrógeno-yodo
Para la reacción
H 2 + I 2 → 2HI(22-19)la ecuación de tasa observada es
![-\ frac {d [H_2]} {dt}\, =k [H_2] [I_2]](https://upload.wikimedia.org/math/1/0/9/10936ca155d79223d8b8db7d0c7f1fb7.png) (22-20)
(22-20)Sin embargo, tanto N. N. Semenov como Henry Eyring han sugerido que el verdadero mecanismo podría no ser el de la ecuación 22-19, sino un mecanismo de dos etapas que implica la disociación reversible de I 2 a 2I, seguido de la reacción trimolecular de I y H 2:
I 2 ↔ 2I
H 2 + 2I → 2HI(22-21)La velocidad de expresión para la reacción de una molécula H 2 con dos átomos I es
![\ frac {d [H_2]} {dt}\, =k' [H_2] [I] ^ {2}](https://upload.wikimedia.org/math/9/d/b/9dbb9ec0aec24e8144bae5d97d8b492d.png) (22-22)
(22-22)y si la disociación de I 2 es reversible y en equilibrio, podemos escribir una expresión constante de equilibrio:
![K =\ frac {[I] ^2} {[I_2]}\,](https://upload.wikimedia.org/math/a/0/6/a06b3c678b9e6e3f17681b2f70a3f801.png) [I] 2 =K [I 2](22-23)
[I] 2 =K [I 2](22-23)Luego sustituyendo la concentración de átomos I en la ecuación 22-22 por la ecuación de equilibrio 22-23 produce
![-\ frac {d [H_2]} {dt}\, =K'k [H_2] [I_2]](https://upload.wikimedia.org/math/a/9/3/a9328cd04ee1380cde450d3c59900cf9.png) (22-24)
(22-24)que es la misma tasa de expresión que si el mecanismo fuera realmente uno de colisión bimolecular. Por lo tanto, tenemos dos mecanismos diferentes con la misma expresión de tasa. ¿Cómo podemos elegir entre ellos?. (Por encima de 800 K, ocurren reacciones secundarias con diferentes mecanismos, sin embargo, estas reacciones pueden descuidarse a temperaturas moderadas).
Los dos mecanismos tienen la misma ecuación de velocidad siempre que la disociación de I2 esté en equilibrio térmico, y la cantidad de átomos I presentes viene dada por la constante de equilibrio térmico de la ecuación 22-23. A temperaturas más altas, más I 2 se disocia, produciendo así el mismo efecto que habría resultado de la mayor constante de velocidad bimolecular en el mecanismo bimolecular. J. H. Sullivan decidió probar los dos mecanismos haciendo que la concentración de átomos de yodo fuera diferente de lo que normalmente es en la disociación térmica de I 2. Lo hizo disociando I 2 con luz de 578-nm de una lámpara de vapor de mercurio. Esta luz debería haber tenido poco efecto si la reacción fue bimolecular, aparte de una ligera disminución en la concentración de I 2. Por el contrario, si la reacción trimolecular fue correcta, la velocidad de reacción debería haber aumentado con la intensidad de la luz irradiante ya que se estaban produciendo más átomos de I.
Sullivan calculó la concentración de átomos I presentes en varias intensidades de luz irradiante y encontró que la tasa de aparición de HI es proporcional al cuadrado de la concentración de átomos I. Por lo tanto, el mecanismo de la ecuación 22-21 es el correcto. La reacción clásica H 2 + I 2 es una reacción trimolecular que imita una reacción bimolecular más simple debido al equilibrio térmico que normalmente existe entre I 2 y 2I. (Al menos, hasta que alguien aún más ingenioso diseñe un experimento que demuestre que es una reacción más complicada imitando una reacción trimolecular). Como señala Sullivan [j Chem. Phys. 46, 73 (1967)], la reacción trimolecular
H 2 +2I→2HIpuede ser reemplazado por dos etapas bimoleculares:
H 2 +2I→H 2 I
H 2 I+I 2 ↔ HISi el primero de ellos es rápido y reversible, de manera que los reactivos y los productos están en equilibrio, entonces la expresión de la velocidad es la misma que para el proceso trimolecular, y los dos mecanismos no pueden distinguirse por las velocidades de reacción. Este ejemplo hace un punto que hay que tener presente en todo momento -nunca podremos probar que un mecanismo propuesto es correcto; sólo podemos probar que aún no se ha demostrado que esté equivocado. Siempre existe la posibilidad de que un experimento más sutil, como la perturbación de Sullivan del equilibrio térmico con la luz, pueda descubrir las debilidades en un mecanismo aceptado. Cuando se presentan dos teorías sobre un mismo tema, la tentación (y generalmente la elección más sabia) es optar por la más simple, hasta que los datos te obliguen a hacer lo contrario. Pero siempre debes estar preparado para cambiar de opinión cuando los nuevos datos lo demanden.
Tasas y Mecanismos de Reacciones de Sustitución
La reacción del bromuro de terc-butilo con OH —,
(CH 3) 3 CbR + OH - ↔ (CH 3) 3 COH + Br -(22-25)tiene la expresión de velocidad experimental
-![\ frac {d [(CH_3) _3cBr]} {dt}\, =k [(CH_3) _3cBr]](https://upload.wikimedia.org/math/6/e/1/6e1522fef9751ac914ba5ebf40bab96a.png) (22-26)
(22-26)La tasa no parece depender en absoluto de la concentración de OH -. Por el contrario, la reacción similar con un átomo de carbono menos altamente sustituido en bromuro de etilo,
CH 3 CH 2 Br + OH - ↔ CH 3 CH 2 OH + Br(22-27)tiene la expresión de velocidad que podríamos esperar de la ecuación química:
![-\ frac {d [ch_3ch_2br]} {dt}\, =k [CH_3ch_2br] [OH^-]](https://upload.wikimedia.org/math/f/2/f/f2f830c15fe292e97817d76a37fd913f.png) (22-28)
(22-28)¿Por qué estas dos reacciones similares deberían proceder por diferentes mecanismos y tener diferentes ecuaciones de velocidad? ¿Y cómo es que la tasa en la ecuación 22-26 puede ser independiente de la concentración de uno de los reactivos?
La reacción 22-25 se lleva a cabo por el mecanismo S N 1 la ecuación

El bromuro de terc-butilo primero se disocia en una reacción lenta, y luego el ion carbonio que se forma reacciona inmediatamente con OH —. Siempre que un proceso se lleva a cabo mediante una serie de pasos rápidos con un paso más lento, la velocidad general de reacción será controlada por la etapa lenta. La velocidad en esta reacción de S N 1 depende completamente de la rapidez con la que se descomponen las moléculas de (CH 3) 3 CbR. La capacidad de hacer reaccionar iones carbonio con OH — supera con creces la cantidad de iones carbonio suministrada por la disociación del bromuro de terc-butilo. La cantidad total de OH - presente no es importante.
¿Por qué las reacciones van con diferentes mecanismos? El esquema ThES N 2 es posible para el bromuro de etilo porque hay espacio para tres sustituyentes del átomo de C (CH 3 y dos H), más OH — y Br. Debido a esto el complejo activado
-
es estericamente posible.
En el bromuro de terc-butilo, C 4 H 9 Br, por otro lado, los grupos unidos al átomo de carbono (tres CH 3) son lo suficientemente grandes como para que OH -y Br - no puedan unirse al mismo tiempo. El complejo activado de la reacción de S N 2 es imposible, y ninguna reacción puede ocurrir hasta que una molécula de bromuro de terc-butilo se disocie espontáneamente. El Ion Carbonio disociado es entonces el ion a atacar, ya sea por Br - para volver a formar los reactivos o por OH — para formar el producto. Si Br —está presente solo como resultado de una reacción previa de bromuro de terc-butilo, su concentración es probablemente mucho menor que la del OH —, y la mayoría de los iones carbonio se convertirán en alcohol terc-butílico, (CH 3) 3 COH.
En general, una expresión de velocidad que no está de acuerdo con la estequiometría de la reacción global es una indicación de que la reacción avanza por una serie de etapas. Entonces el problema es encontrar un conjunto de pasos incluyendo un paso lento que dé cuenta de la ley de tasas observada.
En los mecanismos de reacción del bromuro de terc-butilo y bromuro de etilo la diferencia de velocidad también se encuentra en el plano octaédrico y cuadrado
complejos de los metales de transición. Los complejos planos cuadrados de Pt (II) y otros metales pueden reaccionar con nuevos ligandos por mecanismos asociativos (S N 2) porque el átomo metálico es accesible desde ambos lados del plano. El mecanismo S N 2 de la reacción
Pt (NH 3) 3 Cl + Br - → Pt (NH 3) 3 Br+ + Clse puede escribir
-
El complejo activado es un platino de cinco coordinados, que se descompone rápidamente en productos. La velocidad de la reacción global depende de la velocidad de formación del complejo activado. Esta tasa está fuertemente influenciada por la naturaleza del grupo entrante (Br - en este ejemplo). Los ligandos capaces de formar enlaces fuertes con el átomo central son los grupos que mejor entran porque desplazan rápidamente al grupo lábil (C1 en este ejemplo). Los iones CN —e I - son buenos grupos de entrada para los complejos de Pt (II), mientras que NH 3 y H 2 0 son relativamente pobres.
Es mucho más difícil que los complejos octaédricos de seis coordinados reaccionen por un mecanismo S N 2 porque seis ligandos alrededor de un metal central, como el Co (III), dejan poco o ningún espacio para la unión de un grupo entrante en estado de transición. Estudios de reacciones de sustitución de complejos octaédricos de Co (III) han establecido que la etapa determinante de la velocidad implica la disociación del enlace entre el Co (III) y el grupo saliente (el grupo entrante no está involucrado en esta etapa de disociación). En solución acuosa, por ejemplo, H 2 0 desplaza Cl - en el complejo Co (NH 3) 5 Cl 2+, produciendo así Co (NH 3) 5 H 2 0 3+. El mecanismo que es consistente con la velocidad experimental de esta y similares reacciones es el mecanismo disociativo o S N 1, que puede escribirse.
- [
Para tales mecanismos, el grupo entrante no juega un papel significativo en la creación del estado de Transición y la velocidad de la reacción general. Una característica de la mayoría de las reacciones de sustitución octaédrica es la falta de influencia de los grupos que ingresan en la velocidad de reacción.
Reacciones en Cadena
La reacción H 2 + Br 2 → 2HBr tiene la extraña ecuación de velocidad que ya hemos visto,
(22-3)Durante 13 años después de que se descubrió esta ley de tasas, nadie pudo contabilizarla. Entonces, tres grupos científicos esos grupos de Henry Eyring, K. F. Herzfeld, y Michael Polanyidid así simultáneamente. Propusieron que la reacción procede por un mecanismo de cadena que implica dos etapas de propagación de cadena:
(1) H 2 +Br→ H+HBr (k 1)
(2) H+ Br 2 → HBr+Br (k 2)Cuando una molécula se rompe en fragmentos no cargados que tienen electrones desapareados, los electrones desapareados (por ejemplo, en H y Br) hacen que los fragmentos llamados radicales sean químicamente reactivos. El producto atómico de cada una de estas etapas es un reactivo para el otro paso, y ambos producen HBr. Así, HBr resulta, no de una colisión bimolecular, sino de una cadena interminable de reacciones 1 y 2. El primero de estos dos pasos es la reacción de la Figura 22-9. Pero, ¿de dónde vienen estos átomos de Br y H? Se postula que los átomos Br provienen inicialmente de una etapa de inicio de cadena:
(3) Br 2 → Br+ Br (k 3)¿Por qué no se incluye también la disociación de H 2? La verdadera razón es que la explicación de la velocidad de reacción (ecuación 22-3) no la requiere, y si la agregamos, obtenemos la expresión de velocidad incorrecta. Podemos justificar esta omisión de otra manera: La energía de disociación de H 2 es 432 kJ mol -1, mientras que la de Br 2 es solo 190 kJ mol -1. Una gran concentración de HBr inhibe la reacción, como podemos ver en el término HBr en el denominador de la ecuación 22-3, y una gran concentración de Br 2 contrarresta esta inhibición. De esto es evidente que HBr y Br 2 están compitiendo por la misma sustancia química. ¿Cuál podría ser esa sustancia? El candidato más probable son los átomos de hidrógeno y la reacción inhibidora sería
(4) H+HBr→h 2 +Br (k 4)Esta es una reacción inhibidora de cadena y se contrarresta si un exceso de Br 2 hace que la reacción 2 vaya rápidamente, como predice la ecuación de velocidad 22-3. Finalmente, la cadena se termina por la recombinación de Br:
(5) Br+Br → Br 2 (k 5)Si podemos obtener la ecuación 22-3 a partir de estas cinco reacciones este será un argumento fuerte para la corrección de este mecanismo de cadena (aunque no una prueba absoluta, como hemos visto con HI.) La tasa de aparición de HBr viene dada por
![\ frac {d [HBr]} {dt}\, =+k_1 [H_2] [Br] +k_2 [H] [Br_2] -k_4 [H] [HBr]](https://upload.wikimedia.org/math/9/d/c/9dc0f8d966369a973e1bf809c64b8b96.png) (22-29)
(22-29)ya que HBr aparece como resultado de las reacciones 1 y 2, y desaparece en la reacción 4. Las tasas de producción de átomos de H y Br vienen dadas por
![\ frac {d [H]} {dt}\, =k_1 [H_2] [Br] +k_2 [H] [Br_2] -k_4 [H] [HBr]](https://upload.wikimedia.org/math/1/c/4/1c42410e7198aadfe94d95fc9dcdb7ab.png) (22-30)
(22-30)![\ frac {d [Br]} {dt}\, =-k_1 [H_2] [Br] +k_2 [H] [Br_2] +k_4 [H] [HBr] +2k_3 [Br_2] -2k_5 [Br] ^ {2}](https://upload.wikimedia.org/math/9/1/0/910864e2d58637589bc8d73ddf95bab8.png) (22-31)
(22-31)Los coeficientes de 2 frente a k 3 y k 5 surgen porque cada unidad de reacción 3 produce dos átomos de Br, y cada unidad de reacción 5 elimina dos átomos de Br.
En este punto se debe hacer una simplificación esencial. Se supone que la cantidad real de átomos de H y Br presentes en cualquier momento debe ser pequeña porque se consumen casi a la misma velocidad que se producen. Poco después de que comience la reacción, las concentraciones de H y Br alcanzarán un estado estacionario y permanecerán constantes mientras la reacción continúe con un suministro abundante de reactivos. Debido a esto, cada una de las ecuaciones de tasa en 22-28 y 22-29 se puede establecer igual a cero:
![0=k_1 [H_2] [Br] -k_2 [H] [Br_2] -k_4 [H] [HBr]](https://upload.wikimedia.org/math/8/0/4/8047f6bdb56ee9e0dbb11402d7a2f484.png) (22-32)
(22-32)![0=-k_1 [H_2] [Br] +k_2 [H] [Br_2] +k_4 [H] [HBr] +2k_3 [Br_2] -2k_5 [Br] ^ {2}](https://upload.wikimedia.org/math/8/8/1/881083fe43ef8d65d7531828c6092a43.png) (22-33)
(22-33)Sumando estas dos ecuaciones rinde
![2k_5 [Br] ^ {2} =2k_3 [Br_2]](https://upload.wikimedia.org/math/3/6/3/36316b3c23c305b6c6fee65a973a7419.png)
con la ecuación de tasa
![[Br] = (\ frac {k_3} {k_5}\,) ^ {1/2} [Br_2] ^ {2}](https://upload.wikimedia.org/math/c/1/3/c13696968c26fb4486b9b14c84ac12e3.png) (22-34)
(22-34)Este cálculo nos da una concentración en estado estacionario para los átomos de Br en términos de la concentración de moléculas de Br 2. La ecuación de tasa HBr se puede reescribir como
![\ frac {d [HBr]} {dt}\, =k_1 [H_2] [Br] + (k_2 [Br_2] -k_4 [HBr]) [H]](https://upload.wikimedia.org/math/a/8/b/a8b1c459f5b015b9e5c59b15ed80f2e4.png) (22-35)
(22-35)Podemos eliminar la concentración de H expresándola en términos de concentración de Br de la ecuación 22-32:
![[H] =\ frac {k_1 [H2]} {k_2 [Br_2] +k_4 [HBr]}\, [Br]](https://upload.wikimedia.org/math/2/b/6/2b6ce90e110aaf544013d9532ea0f996.png) (22-36)
(22-36)Sustituir la ecuación 22-36 por 22-35, colocar todo sobre un denominador común y cancelar términos rinde
![\ frac {d [HBr]} {dt}\, =\ frac {2k_1k_2 [H_2] [Br_2] [Br]} {k_2 [Br_2] +k_4 [HBr]}\,](https://upload.wikimedia.org/math/7/a/c/7acdf6a1c9b09c9a04e016d35aa0fb73.png) (22-37)
(22-37)Dividiendo arriba e abajo por [Br2] y luego eliminando [Br] con la ecuación 22-34 rinde
![\ frac {d [HBr]} {dt}\, =\ frac {1} {1+ (k_4/k_2) ([HBr]/[Br_2])}\,](https://upload.wikimedia.org/math/0/8/2/08206436ed10964e8f26e341d763895f.png) * (2k 1 (k 3 /k 5) 1/2 [H 2] [Br 2] 1/2)(22-38)
* (2k 1 (k 3 /k 5) 1/2 [H 2] [Br 2] 1/2)(22-38)Esta es exactamente la ley de velocidad experimental, en la que las constantes de velocidad experimentales están relacionadas con las de las reacciones individuales en la cadena por
k = 2k 1 (k 3 /k 5) 1/2
k'= k 4 /k 2Ahora que sabemos lo que significan estas dos constantes experimentales en términos de las reacciones individuales, podemos dar una interpretación mucho más completa a la ley de tasas, ecuación 22-38. Supongamos que podríamos variar las constantes de tasa individuales, k 1 a k 3, a voluntad. ¿Qué efectos tendrían estos cambios en la tasa general? La tasa general de producción de HBr se acelera si las constantes de velocidad k 1, k 2 y k 3 son grandes, o si las reacciones 1, 2 y 3 son rápidas. Las dos primeras de estas reacciones producen HBr; la tercera prepara el camino haciendo más átomos de Br. La producción de HBr se ralentiza si k 4 y k 5 son grandes, o si las reacciones inhibidoras de cadena y terminación de cadena son rápidas.
Mientras k 3 y k 5 cambien juntos, no hay cambio en la velocidad general de reacción. Las reacciones 3 y 5 son las etapas opuestas de inicio y terminación. De igual manera, siempre y cuando k 2 y k 4 cambien juntos, la tasa no se ve afectada. Esto, también, es sensible; para las reacciones 2 y 4 son similares en que ambas consumen un H y producen un Br, pero difieren en producir HBr en la reacción 2 y eliminarlo en la reacción 4. La inhibición por HBr ocurre porque la reacción 4 está potenciada, y la inhibición es disminuida por Br 2 porque la reacción 2 está potenciada.Usos prácticos de las Reacciones Complejas
Aunque la química detrás de la reacción compleja no siempre se entiende, ni siquiera se piensa en ella, estas reacciones se utilizan muy a menudo en el mundo que nos rodea.
Un excelente ejemplo de esto es el yoduro de hidrógeno que se utiliza como reactivo en la reacción compleja que resulta en la producción de metanfetamina. La producción de metanfetamina utiliza la reducción de efedrina con fósforo rojo y yoduro de hidrógeno (también conocido como ácido hidriódico) [5]. El mecanismo de reacción para la reducción de la efedrina con fósforo rojo y yoduro de hidrógeno se resume de la siguiente manera: La efedrina reacciona con yoduro de hidrógeno para formar yodoefedrina (yodometanfetamina) que luego se reduce a metanfetamina [6]. La reducción de efedrina con fósforo rojo y yoduro de hidrógeno para crear metanfetamina implica una oxidación cíclica del anión yoduro a yodo y la reducción del yodo de nuevo al anión por el fósforo rojo [7]. Este proceso de múltiples etapas es una reacción compleja clásica en la que dos reactivos experimentan una reacción de oxidación a partir de un producto intermedio. Este producto químico intermedio experimenta una reacción de reducción con un tercer químico, y transforma uno de los reactivos originales de nuevo a su estado original además de cambiar el intermedio al producto final.
Clásicamente, la metanfetamina ha sido considerada como una sustancia ilícita que utiliza productos químicos agresivos para liberar una cascada de dopamina del cerebro [8]. Si bien es cierto que la metanfetamina puede ser abusada de esta manera, la metanfetamina también se usa comúnmente en medicina también. En dosis controladas, y en pastillas de liberación temporal especialmente diseñadas, la metanfetamina y la anfetamina se utilizan para tratar afecciones como el Trastorno por Déficit de Atención con Hiperactividad, Lesión Cerebral Traumática, y los síntomas de somnolencia diurna de Narcolepsia y Síndrome de Fatiga Crónica. [9]. Una dosis diaria típica de metanfetamina oral para el tratamiento del trastorno por déficit de atención e hiperactividad en niños es de 20—25 mg. En el cerebro la metanfetamina eleva los niveles de neurotransmisores extracelulares monoamínicos (dopamina, serotonina, norepinefrina) al promover su liberación de las terminaciones nerviosas. No se entiende completamente cómo la metanfetamina provoca la liberación de neurotransmisores, pero parece implicar cambiar la distribución de los neurotransmisores monoamínicos de las vesículas sinápticas al citoplasma neuronal. También invierte el transporte de neurotransmisores desde dentro de la membrana plasmática celular hacia el espacio extracelular [10]. Este cambio en la química corporal da como resultado los siguientes síntomas y efectos: estado de alerta, vigilia, energía, bienestar, euforia (a dosis altas) y supresión del apetito. La metanfetamina también activa el sistema cardiovascular (aumento de la frecuencia cardíaca y la presión arterial) y, por esta razón, puede causar la muerte a dosis altas [11].
Otra forma en la que se utilizan las reacciones complejas, y más específicamente las reacciones en cadena, es en las reacciones de fisión nuclear. En estas reacciones un átomo es bombardeado por un neutrón que divide el átomo en dos átomos que tienen un peso combinado menor que el del átomo original. [12] Esta reacción libera varios productos: 1) dos átomos más pequeños; 2) calor; y 3) más neutrones. Esto permite que la reacción no sólo continúe, sino que crezca en tamaño a escala exponencial, convirtiéndose así en una reacción en cadena. Este es el concepto detrás de los reactores nucleares y las explosiones nucleares; una reacción en cadena nuclear sostenida de moléculas que produce energía. Para que esta reacción ocurra y sea eficiente, deben usarse átomos de alto peso molecular, como el plutonio y el uranio, debido a que su Energía de Unión es baja. En consecuencia, cuando se divide el átomo, la cantidad de energía liberada es grande. También hay una masa específica, conocida como la masa crítica, que es la masa que los reactivos deben ser acumulativamente para que la reacción siga siendo sustentable. Si se cumplen todos estos criterios entonces se producirá una reacción de fisión y resultará en una liberación de energía. La diferencia entre un reactor de fisión y una bomba de fisión es la velocidad a la que se produce la reacción de fisión. [13] En un reactor nuclear el metal está en un moderador, lo que ralentiza la velocidad de la reacción, y la reacción es dispersada por barras de control que absorben una porción de los neutrones libres y ralentizan aún más la reacción. [14] En estas reacciones de fisión se utiliza el moderador, generalmente agua, para hacer vapor que alimenta una turbina y produce energía eléctrica. [15]
Referencias
- ALSNews, Fotodisociación de ozono sondeada usando luz onduladora, http://www.als.lbl.gov/als/als_news/...35_090595.html
- Gale Group Chemistry:Sondeo dinámica de reacciones, es.wikibooks.org/w/index.php?... editar§ion=5
- Experimento de haz molecular cruzado de Yuan T. Lee, http://www.osti.gov/accomplishments/YuanLee_Exp.pdf
- Walsh R, Martin E, Darvesh S. Un método para describir reacciones catalizadas por enzimas mediante la combinación de parámetros cinéticos enzimáticos en estado estacionario y de curso de tiempo. Biochim Biophys Acta. 2010, 1800 (1), pp. 1-5
- Skinner, Henry F., “Síntesis de metanfetamina a través de HI/Reducción de fósforo rojo de efedrina”, Forensic Science International, 48 128-134 (1990)
- Skinner, Henry F., “Síntesis de metanfetamina a través de HI/Reducción de fósforo rojo de efedrina”, Forensic Science International, 48 128-134 (1990)
- Skinner, Henry F., “Síntesis de metanfetamina a través de HI/Reducción de fósforo rojo de efedrina”, Forensic Science International, 48:128-134 (1990)
- Kish, Stephen J., “Mecanismos farmacológicos de la metanfetamina cristalina”, CMAJ 13:178 (17 de junio de 2008)
- Kish, Stephen J., “Mecanismos farmacológicos de la metanfetamina cristalina”, CMAJ 13:178 (17 de junio de 2008)
- Kish, Stephen J., “Mecanismos farmacológicos de la metanfetamina cristalina”, CMAJ 13:178 (17 de junio de 2008)
- Kish, Stephen J., “Mecanismos farmacológicos de la metanfetamina cristalina”, CMAJ 13:178 (17 de junio de 2008)
- Hill, John W., y Ralph H. Petrucci. La química general un enfoque integrado. Ed. Paul F. Corey. Upper Saddle River, N.J: Prentice Hall, 1999.
- Hill, John W., y Ralph H. Petrucci. La química general un enfoque integrado. Ed. Paul F. Corey. Upper Saddle River, N.J: Prentice Hall, 1999.
- Hill, John W., y Ralph H. Petrucci. La química general un enfoque integrado. Ed. Paul F. Corey. Upper Saddle River, N.J: Prentice Hall, 1999.
-
- La hidroformilación es el proceso de producción de aldehídos a partir de alquenos mediante la adición de un grupo formilo (CHO) y un átomo de hidrógeno a un doble enlace carbono-carbono usando catálisis homogénea. Es particularmente útil porque los aldehídos que se forman pueden convertirse en productos útiles como detergentes y productos químicos especiales. [3]
- Los catalizadores Ziegler-Natta son catalizadores a menudo basados en compuestos de titanio y compuestos organometálicos de aluminio. Se utilizan principalmente para polimerizar 1-alquenos terminales. [4]
- La mayor parte del conocimiento actual sobre catálisis homogénea proviene de estudios anteriores de hidrogenación. La hidrogenación (como se muestra en la siguiente figura) es una reacción química que resulta de unir hidrógeno a compuestos orgánicos mediante el uso de catalizadores.

-
Los catalizadores homogéneos involucrados en esta reacción incluyen el catalizador de Wilkinson, un compuesto a base de rodio y el catalizador de Crabtree, un compuesto a base de iridio. Además, muchas aplicaciones modernas de hidrogenación se pueden encontrar en las industrias petroquímica, farmacéutica y alimentaria. [5]Hidrogenación de carvona
- La activación del enlace carbono-hidrógeno o la activación de CH se pueden definir como una reacción que forma un enlace carbono-hidrógeno. Aunque tradicionalmente no son reactivos, los enlaces CH se pueden formar por coordinación usando un catalizador. Un papel importante de la activación de CH es la capacidad de convertir alcanos baratos y abundantes en valiosos compuestos orgánicos funcionalizados. [6]
- En la inhibición reversible competitiva el sustrato y el inhibidor compiten por el sitio activo de la enzima ya que no pueden unirse a él al mismo tiempo. Este tipo de inhibición puede ser superada por altas concentraciones de sustrato. [14] La inhibición reversible competitiva también puede ocurrir cuando la competencia es por el sitio alostérico en lugar del sitio activo. [15]
- En la inhibición reversible no competitiva, el inhibidor se une al complejo creado por el sustrato que se une al sitio activo de la enzima. Debido a que el sitio activo ya no está disponible, la eficiencia de unión disminuye al igual que la velocidad máxima de la enzima. [16]
- En la inhibición mixta el inhibidor puede unirse a la enzima y a su sustrato simultáneamente. Si bien este efecto puede ser reducido por una alta concentración de sustrato no se puede superar por completo. Este tipo de unión simultánea puede ocurrir en el sitio activo de la enzima, pero generalmente ocurre a través del efecto alostérico (El inhibidor se une a un sitio distinto al sitio activo) que cambia la forma de la enzima y reduce la afinidad del sustrato por el sitio activo. [17]
- La inhibición no competitiva, un tipo de inhibición mixta reversible, reduce la actividad de la enzima sin afectar las capacidades de unión del sustrato. El grado en que se produce la inhibición depende directamente de la concentración del inhibidor. [18] El inhibidor siempre se une a un sitio alostérico. [19]
- Bryant, Charles W. y Karim Nice. “Cómo funcionan los convertidores catalíticos”. http://auto.howstuffworks.com/catalytic-converter2.htm.
- Enciclopedia en línea de Brittanica, “Catálisis Homogénea”, http://www.britannica.com/EBchecked/topic/270491/homogeneous-catalysis
- Wikipedia, “Hidroformilación”, es.wikipedia.org/wiki/Hidroformilación
- Wikipedia, “Catalizadores Ziegler-Natta”, es.wikipedia.org/wiki/Ziegler-Natta
- Wikipeida, “Hidrogenación”, es.wikipedia.org/wiki/Hidrogenación
- Wikipedia, “Activación C-H”, es.wikipedia.org/wiki/C-H_Activation
- R. E. Dickerson, H. B. Gray, y G. P. Haight, Jr., Chemical Principles, 3a edición, The Benjamin/Cummings Publishing Company, Menlo Park, CA, 1979, http://resolver.caltech.edu/CaltechBOOK:1979.001
- R. E. Dickerson, H. B. Gray, y G. P. Haight, Jr., Chemical Principles, 3a edición, The Benjamin/Cummings Publishing Company, Menlo Park, CA, 1979, http://resolver.caltech.edu/CaltechBOOK:1979.001
- Acepta: Tecnologías ambientales avanzadas, “Escala de torre de enfriamiento e inhibidores de corrosión” http://www.accepta.com/cooling-towers-water-treatment/cooling-tower-scale-corrosion-inhibitors.asp
- NCI Cancer Bulletin, “Los inhibidores de la aromatasa llegan a la mayoría de edad”, vol. 4/núm. 10, 6 de marzo de 2007 www.cancer.gov/cancertopics/t... inhibidores0307
- Inhibidores de proteasa: una hoja informativa simple de la Red de Datos de Tratamiento del SIDA, 15 de agosto de 2006 http://atdn.org/simple/protease.html
- Informes del consumidor: Best Buy Drugs www.consumerreports.org/healt... ager_ACEIs.pdf
- Wikipedia, Inhibidor de enzimas, es.wikipedia.org/wiki/Inhibidor de enzimas
- Wikipedia, Inhibidor de enzimas, es.wikipedia.org/wiki/Inhibidor de enzimas
- Wikipedia, Inhibición no competitiva, es.wikipedia.org/wiki/Inhibición no competitiva
- Wikipedia, Inhibidor de enzimas, es.wikipedia.org/wiki/Inhibidor de enzimas
- Wikipedia, Inhibidor enzimático, http://en.Wikipedia.org/wiki/Enzyme_inhibitor
- Wikipedia, Inhibidor enzimático, http://en.Wikipedia.org/wiki/Enzyme_inhibitor
- Wikipedia, Inhibición no competitiva, http://en.Wikipedia.org/wiki/Non-competitive_inhibition
![\ frac {d [C]} {dt}.](https://upload.wikimedia.org/math/c/d/6/cd677dc435be9edaa104d012bee0b8d5.png)
![-\ frac {d [A]} {dt}\, =-\ frac {1} {2}\,\ frac {d [B]} {dt}\, =+\ frac {1} {3}\,\ frac {d [C]} {dt}\,](https://upload.wikimedia.org/math/2/5/b/25bff69019e21da74191a0f29153d384.png)
![\ frac {d [HI]} {dt}\, =-2\ frac {d [H_2]} {dt}\, =-2\ frac {d [I_2]} {dt}\,](https://upload.wikimedia.org/math/9/c/b/9cbda66348aed6ff8755b6963af301ea.png)



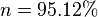




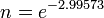




![\ frac {d [NO_2]} {dt} =k [NO] ^2 [O_2]\,](https://upload.wikimedia.org/math/e/1/f/e1f968560ab4a54cf4b8f888ccfd6db0.png)
![-\ frac {d [C_2H_5OH]} {dt} =k [C_2H_5OH] [B_ {10} H_ {14}]\,](https://upload.wikimedia.org/math/f/2/f/f2f01d82787bea7781134ba0efb67082.png)

![\ frac {-d [A]} {dt} = k [A] [B]](https://upload.wikimedia.org/math/4/2/9/4297f466ae4a8089cdf01dc971f61085.png)

![Calidad1\ = k_1 [A] [B]](https://upload.wikimedia.org/math/b/8/0/b808365b50211c183d9cd3061d2e263e.png)
![Calidad2\ = k_2 [C] [D]](https://upload.wikimedia.org/math/5/5/3/553bc1907e46b190cb688bb4654fb22e.png)
![[A] [B] k_1\ = k_2 [C] [D]](https://upload.wikimedia.org/math/e/a/4/ea4d3c9df7377662b36afbcaba8ccf9f.png)
![K_ {eq}\ =\ frac {k_1} {k_2}\ =\ frac {[C] [D]} {[A] [B]}](https://upload.wikimedia.org/math/2/9/6/296d5020f7d7c031d59ab3224ba29dff.png)




![-\ frac {d [A]} {dt} = (Z [A] [B]) (e^ {\ frac {-e_A} {RT}})](https://upload.wikimedia.org/math/3/4/1/3414b62d6f0478486da43c9a8cbb88d2.png)
![-\ frac {d [A]} {dt} =Ze^ {\ frac {-e_a} {RT}} [A] [B]](https://upload.wikimedia.org/math/a/0/a/a0afa047e19560fdb2d3ea4a21b94d31.png)





![K^ {\ Daga} =\ frac {[AB^ {\ Daga]}} {[A] [B]}](https://upload.wikimedia.org/math/5/e/6/5e63b7fe7e5f50207ea4d2445b923a35.png)
![[AB^ {\ Daga}] =K^ {\ Daga} [A] [B]](https://upload.wikimedia.org/math/5/c/d/5cd502fb7d8ecb9e3fd5bcdb4e86daf9.png)

![-\ frac {d [A]} {dt} =\ kappa\ frac {kT} {h} K^ {\ Daga} [A] [B]](https://upload.wikimedia.org/math/7/f/e/7fe5afa5f892270bd6c3d12fcf33b3b4.png)