
7.1: Unión en Compuestos de Coordinación
- Page ID
- 70157

Vinculación en Compuestos de Coordinación
Este capítulo está dedicado a las teorías de vinculación para los compuestos de coordinación. Pensemos primero en, lo que una buena teoría debería ser capaz de hacer en general. La respuesta es, que debería ser capaz de hacer muchas explicaciones correctas para observaciones experimentales basadas en algunas suposiciones, sensatas. Además, debería ser capaz de predecir observaciones experimentales. Cuanto más pueda explicar y predecir la teoría, y cuanto menos suposiciones necesarias, mejor será la teoría. ¿Qué significa esto para una teoría de la vinculación? ¿Qué podría hacer una buena teoría de vinculación para los compuestos de coordinación? Sin duda, debería ser capaz de explicar y predecir el número de enlaces y la forma de una molécula. Además, debería ser capaz de explicar el magnetismo de las moléculas, en particular el dia- y el paramagnetismo. Recuerde, una molécula es diamagnética cuando no tiene electrones desapareados. Es paramagnético cuando hay electrones desapareados. Una molécula diamagnética es repelida por un campo magnético externo. Una molécula paramagnética es atraída por un campo magnético externo. Debe ser capaz de explicar además la estabilidad y reactividad de los complejos, así como las propiedades ópticas de los complejos. Las propiedades ópticas de los compuestos están ligadas a la unión porque están relacionadas con estados electrónicos.
Teoría de los bonos de valencia
Existen esencialmente tres conceptos de unión que se utilizan para describir la unión en los compuestos de coordinación. La primera es la teoría del vínculo de valencia. El concepto de enlace de valencia fue introducido por Linus Pauling en 1931 para explicar el enlace covalente en moléculas de elementos del grupo principal.
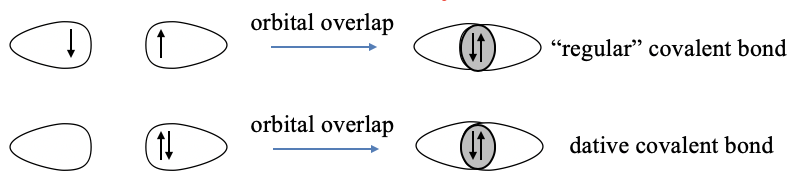
La idea básica es superponer orbitales de valencia semillenos para formar enlaces covalentes en los que los dos electrones se comparten entre los compañeros de unión (Fig. 7.1.1). Estos orbitales pueden ser orbitales atómicos, o orbitales atómicos hibridados. El concepto funciona muy bien para explicar las formas de las moléculas de los elementos del grupo principal. El concepto de enlace de valencia en su forma original asume que cada pareja de enlace aporta un electrón al enlace covalente. Esto no es consistente con el enlace dativo en los compuestos de coordinación donde se asume que un socio dona un par de electrones y el otro socio lo acepta. Para adaptar la teoría de los enlaces de valencia a los compuestos de coordinación, Pauling sugirió que se forma un enlace dativo a través de la superposición de un orbital de valencia completa del donante y un orbital de valencia vacía del aceptor. Veremos que este concepto puede explicar las formas de los compuestos de coordinación en algunos casos, pero en general no funciona muy bien. También veremos que la teoría del enlace de valencia puede explicar el magnetismo en algunos casos, pero también aquí la teoría del enlace de valencia tiene déficits significativos. Por su naturaleza, la teoría del enlace de valencia no puede explicar las propiedades ópticas. En general, la teoría de enlaces de valencia es mucho más adecuada para moléculas de elementos del grupo principal en comparación con los complejos de metales de transición.
Teoría del Campo Cristalino
La segunda teoría principal es la teoría del campo cristalino. En realidad no es una teoría de unión porque se basa en interacciones electrostáticas repulsivas. Originalmente fue desarrollado para explicar el color en los cristales iónicos. Posteriormente, se encontró que también puede explicar colores en compuestos de coordinación molecular, y es adecuado para explicar formas y magnetismo de complejos. Sin embargo, debido a que se basa en interacciones electrostáticas repulsivas, en realidad no puede explicar qué mantiene unidos los átomos en una molécula. Sin embargo, la teoría del campo cristalino es bastante simple y conveniente de usar, y hay mucha practicidad en ella.
Teoría del campo del ligando
La tercera teoría es la teoría del campo de ligandos. Es la teoría más poderosa, pero también la más complicada. Básicamente, es la teoría orbital molecular aplicada a compuestos de coordinación. Puede hacer declaraciones detalladas sobre el número de enlaces y formas de moléculas, y puede explicar el magnetismo y las propiedades ópticas de los compuestos de coordinación.
Teoría de los bonos de valencia para compuestos de coordinación
Complejos Octaédricos
Echemos un vistazo más de cerca a la teoría del vínculo de valencia y evaluemos la teoría de los bonos de valencia para los complejos mediante una serie de ejemplos.
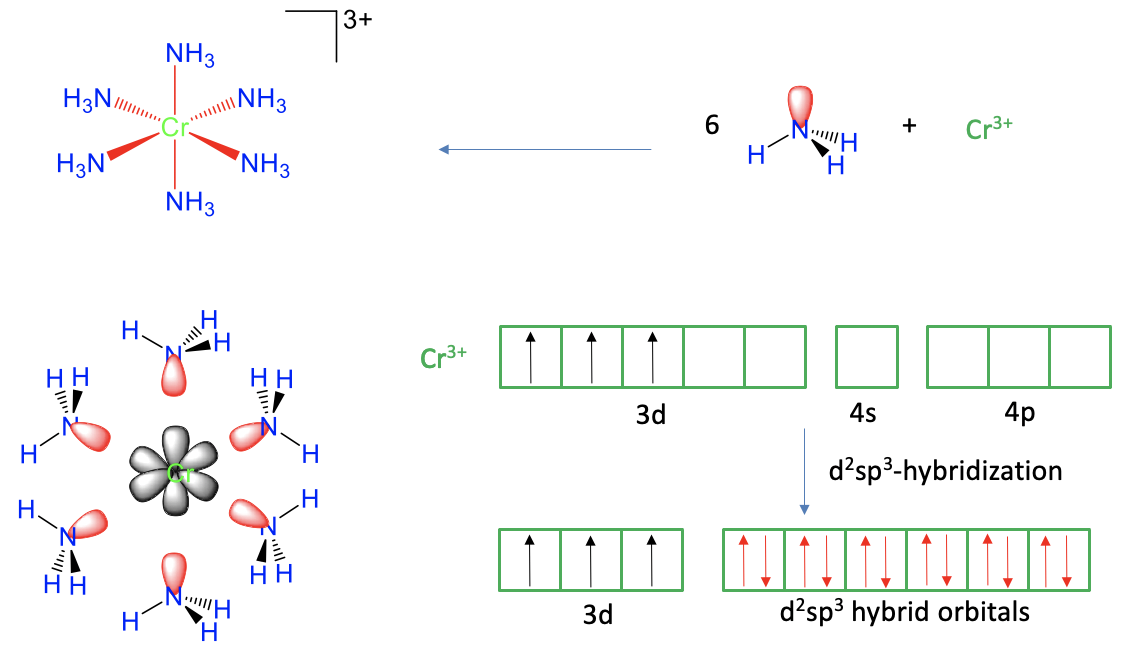 Figura 7.1.2 Teoría del enlace de valencia aplicada al catión hexaamina-cromo (3+)
Figura 7.1.2 Teoría del enlace de valencia aplicada al catión hexaamina-cromo (3+)
El primer ejemplo es el catión hexaammina cromo (3+) (Fig. 7.1.2). Por experimento sabemos que tiene una forma octaédrica, con seis enlaces dativos Cr-N. ¿Puede la teoría de los enlaces de valencia explicar satisfactoriamente los seis enlaces y la forma octaédrica? Para explicar los seis enlaces Cr-N dativos necesitaríamos superponer seis orbitales vacíos de valencia de cromo con seis orbitales de valencia rellenos de N. Podemos ver que los seis ligandos de amina tienen un par solitario de electrones cada uno que puede servir como orbitales de valencia. ¿El cromo tiene seis orbitales de valencia vacíos? Para evaluar esto, primero necesitamos conocer el estado de oxidación del cromo. Es +3 porque los ligandos son todos neutros cuando los enlaces se escinden heterolépticamente, y el catión complejo tiene una carga 3+. Por lo tanto, el cromo es un catión Cr 3 +.
A continuación, necesitamos conocer la configuración electrónica del Cr 3 +. Un átomo de Cr neutro tiene la configuración electrónica 4s 1 3d 5. Cuando un metal de transición pierde electrones para formar un catión, siempre pierde primero sus dos electrones de valencia, y luego sus electrones d. Para el cromo esto significa que debemos eliminar el electrón de un 4s, y dos de los cinco electrones 3d. Se espera que los tres electrones 3d restantes giren en tres orbitales d diferentes de acuerdo con la regla de Hund. ¿Cuántos orbitales de valencia vacíos quedan? Estos serían dos orbitales 3d y los 4s. Además, también estaría justificado considerar los tres orbitales 4p como orbitales de valencia porque los orbitales 4p son energéticamente solo ligeramente más altos que los orbitales 4s. Eso quiere decir que tendríamos los seis orbitales de valencia que necesitaríamos para explicar los seis bonos. Hay, sin embargo, una complicación. Los seis enlaces en el complejo no son distinguibles, pero los seis orbitales de cenefa en el ion Cr 3 + son distinguibles, por ejemplo, los orbitales 3d tienen forma y energía diferentes a los orbitales 4s, que es diferente de los orbitales 4p. Por lo tanto, si superpusiéramos estos orbitales con los pares solitarios de electrones en N, los enlaces no serían equivalentes, ni indistinguibles. Tendríamos dificultades para explicar la forma octaédrica altamente simétrica de la molécula. Para dar la vuelta a este tema, la teoría del vínculo de valencia utiliza el concepto de hibridación. En este concepto mezclamos matemáticamente las funciones de onda de los orbitales de valencia para formar orbitales hibridados. En nuestro ejemplo mezclaríamos los dos orbitales d vacíos, el orbital 4s y los tres orbitales 4p para formar seis orbitales hibridados llamados d 2 sp 3. Tienen la misma forma y tamaño, y sus lóbulos apuntan hacia las esquinas de un octaedro. Por lo tanto, ahora podemos crear superposición entre estos seis orbitales, y los seis pares de electrones solitarios en N para formar seis enlaces Cr-N equivalentes e indistinguibles. Concluimos que ahora hemos explicado satisfactoriamente la vinculación y la forma del complejo.
¿Podemos explicar también su magnetismo? A partir del experimento sabemos que el complejo es paramagnético, y que hay tres electrones desapareados. ¿La teoría de los bonos de valencia predice lo mismo? Sí, lo hace. Hay tres electrones desapareados en los tres orbitales d medio llenos.
Complejos Tetraédricos
Nuestro siguiente es el ejemplo es un complejo tetraédrico, el anión del complejo tetrahidroxo zincato (2-), Fig. 7.1.3.
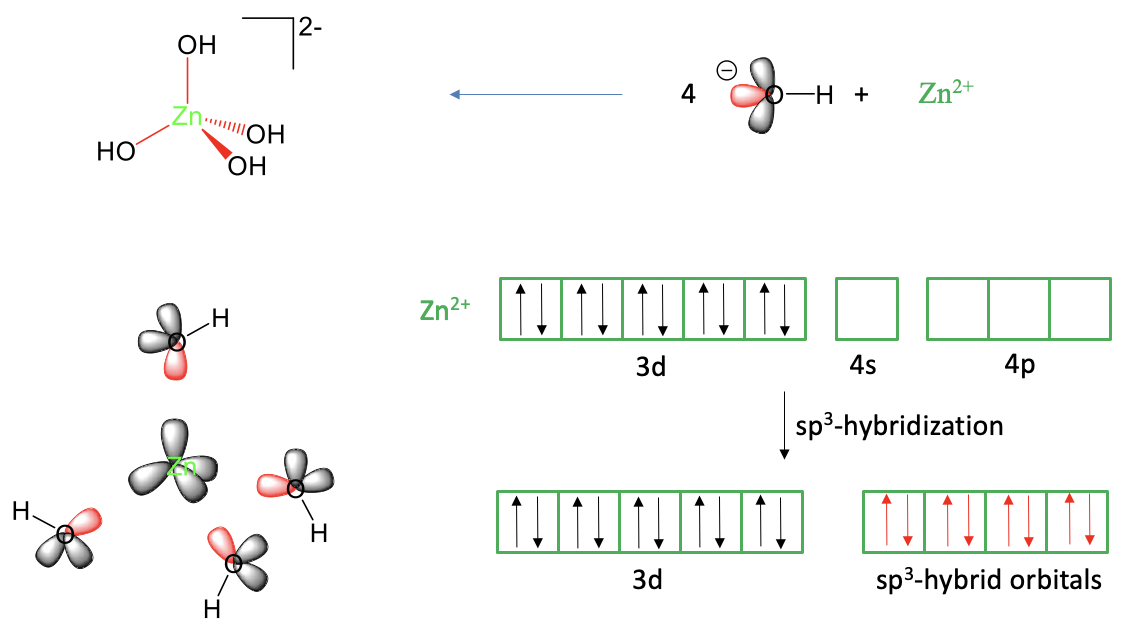
Al verlo como un complejo Lewis-ácido base con enlaces dativos se puede pensar como un aducto de un Zn 2 + y cuatro aniones hidróxido. Uno de los tres pares solitarios de electrones en los iones hidróxido donaría sus electrones en orbitales vacíos de valencia de Zn. Eso significa que necesitaríamos en general cuatro orbitales vacíos de valencia de Zn para explicar los cuatro enlaces Zn-O. Un átomo de zinc neutro tiene la configuración electrónica 4s 2 3d 10. Esto lo podemos derivar del hecho de que el zinc se encuentra en el grupo 12 del periodo 4 en la tabla periódica. Un ion Zn 2 + tiene dos electrones menos. Debido a que debemos eliminar s electrones antes de eliminar d electrones, el Zn 2 + tiene la configuración de electrones 3d 10. Al igual que en el ejemplo anterior podemos considerar justificadamente los orbitales 4p como orbitales de valencia adicionales. Podemos ver que tenemos cuatro orbitales vacíos disponibles para hacer los cuatro enlaces, es decir, los orbitales 4s y 4p, pero estos orbitales no son equivalentes, y no tienen la orientación correcta para explicar la forma tetraédrica del complejo. Hay un ángulo de 90° entre los orbitales p que es menor que el ángulo de enlace tetraédrico de 109.5° en la molécula. Sin embargo, podemos resolver este problema hibridando los orbitales 4s y los tres 4p para formar cuatro orbitales hibridados sp 3. Estos orbitales híbridos tienen la propiedad de que sus lóbulos apuntan hacia las esquinas de un tetraedro. Así, son adecuados para explicar la forma tetraédrica de la molécula. Podemos colocar los ligandos alrededor del ion Zn 2 + y acercarse a los ligandos en los ejes de enlace para crear un solapamiento orbital entre los orbitales hibridados sp 3 vacíos y un par solitario de electrones en el átomo de oxígeno. Esto produce el anión tetrahidroxo cincato (2-) tetraédrico.
¿Podemos explicar también el magnetismo de la molécula? ¿Qué magnetismo predeciría la teoría de los enlaces de valencia? Podemos ver que no hay electrones desapareados en ninguno de los orbitales de valencia metálica. Así, el complejo debe ser diamagnético. Esto es también lo que encontramos experimentalmente. Así, la teoría de los enlaces de valencia es capaz de explicar el magnetismo de este anión complejo.
Complejo Plano Cuadrado
Ahora veamos si la teoría del enlace de valencia también puede explicar un complejo plano cuadrado como el tetracyanonickelato (2-).
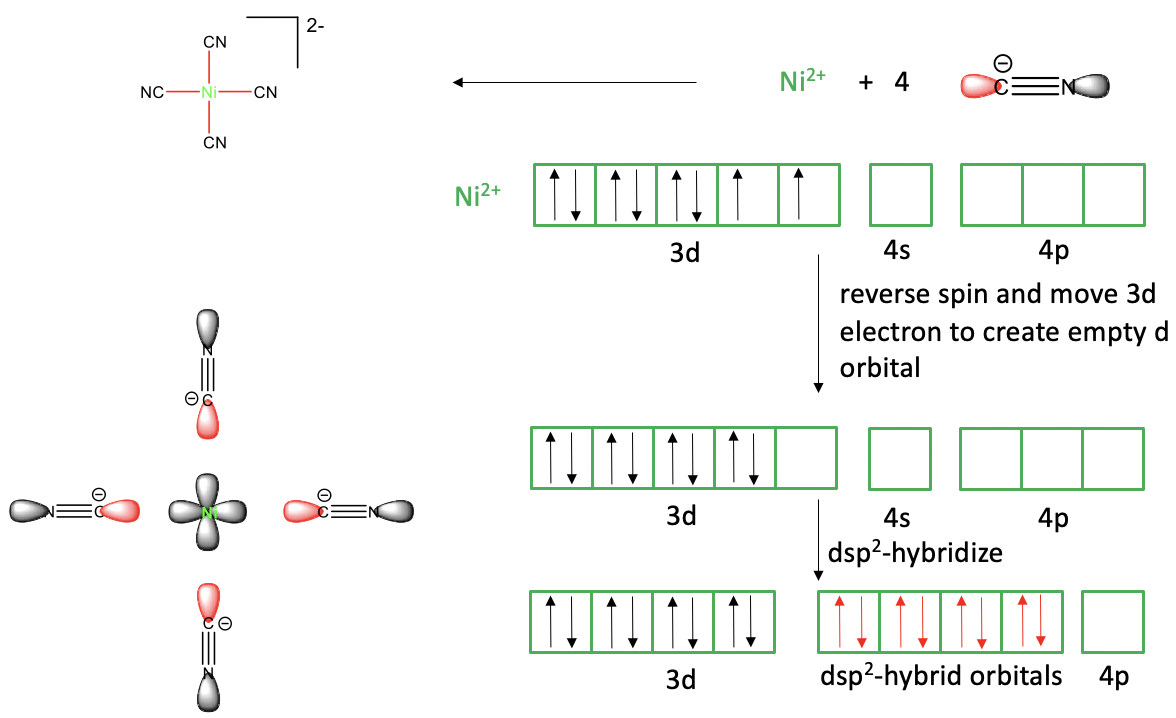 Figura 7.1.4 Teoría del enlace de valencia aplicada al complejo plano de tetracyanonickelato (2-) cuadrado
Figura 7.1.4 Teoría del enlace de valencia aplicada al complejo plano de tetracyanonickelato (2-) cuadrado
En el cuadro de vínculo de valencia vemos los enlaces Ni-CN como enlaces dativos, y el complejo se considera un aducto de Ni 2 + y CN -. Para explicar los cuatro enlaces, el ion Ni 2 + necesitaría tener cuatro orbitales de valencia vacíos. Ni es un metal del grupo 10 y un átomo de Ni neutro tiene la configuración electrónica 4s 2 3d 8. Para crear un ion Ni 2 + debemos eliminar los dos electrones 4s, y así el Ni 2 + tiene la configuración electrónica 3d 8. ¿Tenemos disponibles cuatro orbitales vacíos? Sí, los orbitales 4s y los tres 4p están vacíos pero nuevamente no son equivalentes y por lo tanto no son adecuados para explicar cuatro enlaces Ni-C equivalentes. ¿Podemos hibridar estos orbitales? Sí, podemos, pero los cuatro orbitales hibridados sp 3 resultantes no serían adecuados para explicar la forma plana cuadrada, solo la forma tetraédrica. Lo que sugiere la teoría del enlace de valencia en este caso es revertir el giro de uno de los electrones d desapareados y moverlo al otro orbital d medio lleno. Esto produce un orbital d vacío que ahora podemos hibridar con los orbitales 4s y dos de los 2p a cuatro orbitales hibridados con dsp 2. Estos cuatro orbitales tienen la propiedad de que sus lóbulos apuntan hacia los vértices de un cuadrado, por lo que son adecuados para explicar la forma cuadrado-planar. Podemos acercarnos a los ligandos ahora en los ejes de enlace para crear un solapamiento orbital entre el dsp 2 Ni vacío y los pares de electrones solitarios de los ligandos. También podemos decir que los ligandos donan sus pares de electrones solitarios a los orbitales metálicos hibridados. Esto produce los cuatro enlaces covalentes que necesitamos y produce una molécula de forma plana cuadrada.
Podemos ver que la teoría del enlace de valencia todavía puede explicar la forma plana cuadrada, pero solo con la ayuda de la suposición adicional de que uno de los electrones d obtiene giro invertido y se mueve hacia otro d-orbital. Una suposición que hace una teoría siempre debe ser razonable, así que critiquemos cuán razonable es esta suposición. En primer lugar, ¿es razonable la inversión de giro? La inversión de giro es un proceso quantomecánicamente prohibido, y por lo tanto es cuestionable asumir que sucede. En segundo lugar, no hay una buena explicación de por qué se mueve el electrón. La energía de dos electrones de espín emparejado en el mismo orbital es en realidad más alta que la de dos electrones de espín emparejado en diferentes orbitales. Entonces, en general, vemos que la teoría del vínculo de valencia tiene dificultades para explicar la forma plana cuadrada. Debe hacer suposiciones que no sean muy plausibles.
Complejo orbital octaédrico d 5 de alto y bajo espín
La teoría del enlace de valencia también tiene dificultades para explicar los llamados complejos octaédricos de alto espín y bajo espín. Por ejemplo, se sabe a partir de mediciones magnéticas para iones de metal de transición 3d 5 que pueden hacer complejos octaédricos ya sea con un electrón desapareado o con cinco electrones desapareados, dependiendo del ligando. En el primer caso, se maximiza el número de electrones emparejados en los orbitales d, y tenemos un complejo de espín bajo, en el otro caso se maximiza el número de electrones desapareados, y tenemos un complejo de espín alto. ¿Qué enfoque toma la teoría de los bonos de valencia para explicar este fenómeno?
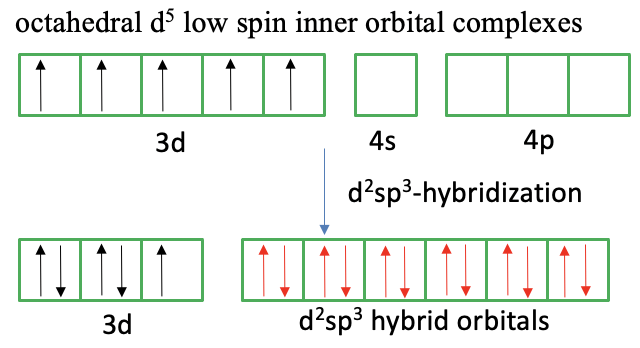
En el caso de un complejo de espín bajo, la teoría del enlace de valencia asume un llamado complejo orbital interno. Al igual que en el complejo plano cuadrado se supone que los electrones desapareados invierten sus espines y se mueven hacia otros orbitales d medio llenos para que se maximice el emparejamiento de espinas. En el caso de un ion d 5, dos electrones invierten su espín y se mueven hacia otros dos orbitales medio llenos. Esto deja un electrón desapareado. Vemos que debido al movimiento de los dos electrones dos orbitales 3D están vacíos ahora, y también lo están los orbitales 4s y los 4p. Los seis orbitales vacíos ahora se pueden combinar para formar orbitales hibridados d 2 sp 3 que pueden explicar las formas octaédricas. Al acercarse a los ligandos se superpone el par solitario de electrones en el ligando con los orbitales híbridos vacíos para formar un enlace covalente dativo. También podemos decir que los ligandos donan pares solitarios de electrones para formar seis enlaces covalentes. Podemos volver a criticar que la inversión de espín está prohibida y el emparejamiento de espinas es energéticamente desfavorable, lo que hace que el enfoque que toma la teoría del vínculo de valencia para explicar el complejo de bajo giro sea insatisfactorio.
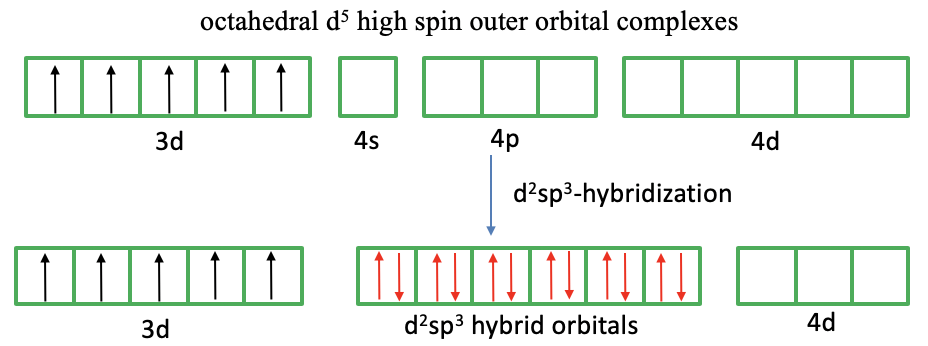
¿Qué pasa con el complejo 3d 5 high spin (Fig. 7.1.6)? En este caso no podemos emparejar giros para crear orbitales d vacíos porque necesitamos explicar cinco electrones desapareados. Ahora, la teoría del vínculo de valencia hace otra nueva suposición. Se supone que los orbitales exteriores 4d se involucran en la vinculación. Estos orbitales están vacíos y disponibles para la hibridación. Por lo tanto, podemos hibridar dos orbitales 4d, 4s y tres 4p para formar orbitales hibridados d 2 sp 3. En el último paso podemos acercarnos a los ligandos, y los ligandos pueden donar sus pares solitarios de electrones a los orbitales d de metales de transición. Ahora hemos explicado los seis enlaces, la forma octaédrica, y los cinco electrones desapareados.
Podemos volver a criticar el enfoque del vínculo de valencia. Qué justificación hay para suponer que los orbitales 4d están involucrados. La respuesta es: Muy poco. Estos orbitales son simplemente demasiado altos en energía para ser considerados orbitales de valencia. No es razonable suponer que están involucrados en la vinculación. Por lo tanto, nuevamente, vemos que la teoría del vínculo de valencia tiene dificultades para explicar las propiedades de un complejo. La teoría del vínculo de valencia tampoco explica las distorsiones de los complejos octaédricos debido al efecto Jahn-Teller.
Complejo orbital octaédrico d 7 de alto y bajo espín
Los complejos de alto y bajo espín no solo se observan para los complejos octaédricos de iones d 5, sino también para los complejos de iones d 7 octaédricos. Un complejo de espín bajo tiene tres electrones desapareados y un complejo de espín alto tiene un electrón desapareado. Veremos que la teoría de los bonos de valencia tiene problemas aún mayores para explicar estos compuestos.
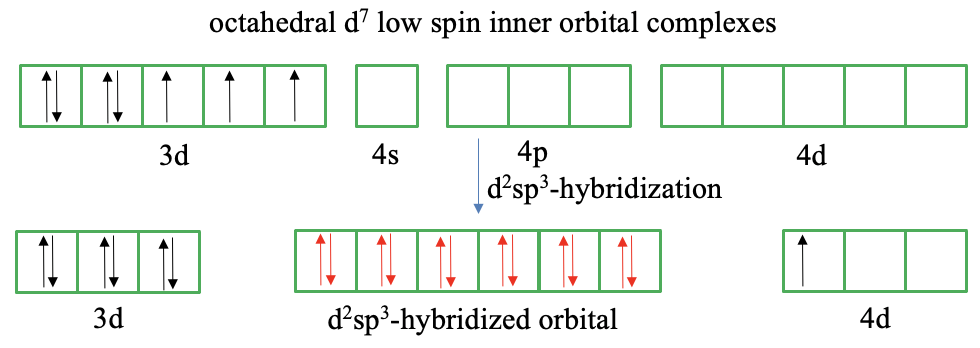
En un ion d 7 hay cuatro electrones emparejados y tres desapareados de acuerdo con la regla de Hund (Fig. 7.1.7). Podemos revertir el espín de un electrón desapareado y emparejarlo con un electrón desapareado en otro orbital medio lleno para reducir el número de electrones desapareados a uno. Sin embargo, esto nos da solo un orbital 3d vacío disponible para la hibridación d 2 sp 3. En este caso no podemos producir orbitales 3D más vacíos invirtiendo el giro. Por lo tanto, debemos hacer de nuevo la cuestionable suposición de que los orbitales exteriores están involucrados en la vinculación como los orbitales 4d. La teoría del enlace de valencia sugiere ahora mover el electrón desapareado del orbital 3d al 4d. Esto se hace simplemente para crear otro orbital 3d vacío que necesitamos para la hibridación d 2 sp 3. Sin embargo, ¿por qué el electrón 3d simplemente entraría en otro orbital de energía mucho mayor? Sin embargo, si hacemos esta suposición cuestionable, efectivamente tenemos seis orbitales disponibles para la hibridación, y podemos dejar que los ligandos donen un par de electrones a los orbitales híbridos vacíos.
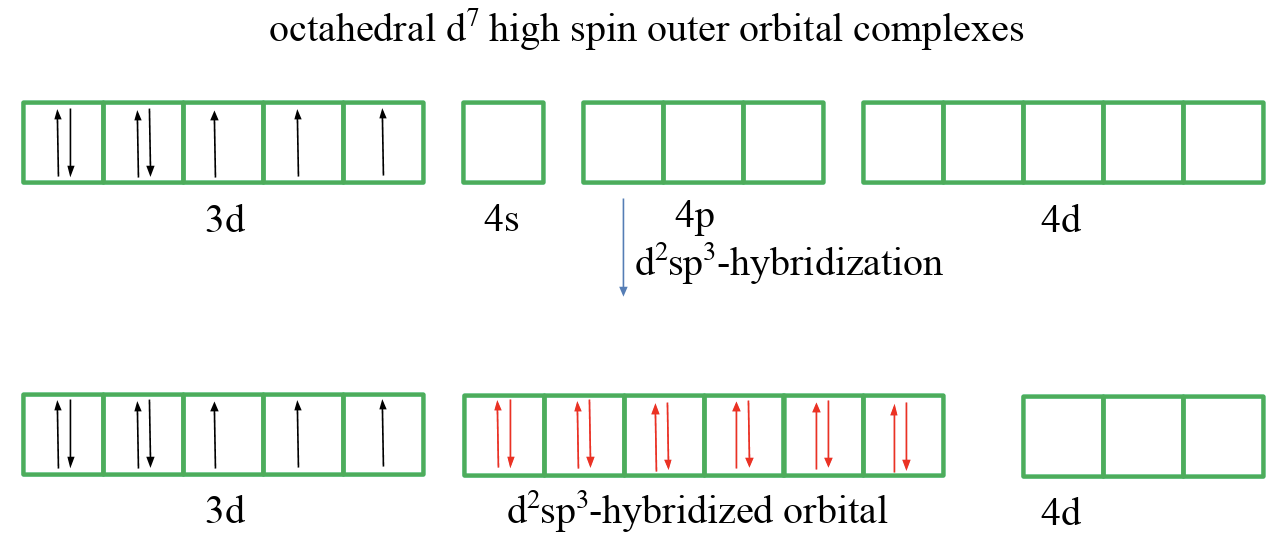
Finalmente, discutamos un complejo orbital externo octaédrico d 7 de alto espín (Fig. 7.1.8). En este caso no podemos emparejar ningún giro en los orbitales 3d. Por lo tanto asumimos nuevamente que los orbitales 4d se involucran en la vinculación, e hibridan dos de ellos con los orbitales 4s y los tres 4p. Los seis ligandos pueden entonces donar seis pares de electrones a los orbitales creando así seis enlaces y explicando la forma octaédrica.
En general, vemos que en la teoría de los enlaces de valencia nos movemos alrededor de los electrones como nos plazca para explicar las formas y el magnetismo de los complejos sin una buena justificación. Por lo tanto, la teoría de los enlaces de valencia, aunque extremadamente valiosa para los compuestos del grupo principal, solo es de uso limitado para los complejos de metales de transición.
Teoría del Campo Cristalino
Ahora vamos a discutir la segunda teoría de vinculación para los compuestos de coordinación, la teoría del campo cristalino. En realidad no es una teoría de unión porque se basa en interacciones electrostáticas repulsivas. Sin embargo, tiene muchas características de una teoría de unión en el sentido de que puede explicar muchos fenómenos que una unión puede explicar, en particular la forma molecular, el magnetismo y las propiedades ópticas.
¿Cuáles son los principios de la teoría del campo cristalino? La teoría del campo cristalino asume que los electrones en los d-orbitales metálicos están rodeados por un campo eléctrico que es causado por los electrones ligandos. A este campo eléctrico se le llama campo de cristal. El nombre campo cristalino proviene del hecho de que este principio se aplicó primero a iones de metales de transición rodeados de aniones en cristales, y solo más tarde se extendió a iones de metales de transición rodeados por ligandos en compuestos de coordinación molecular. La suposición de que los ligandos que rodean un ion de metal de transición producen un campo eléctrico tiene sentido porque los ligandos contienen electrones que están asociados con un campo eléctrico. Se supone además que el campo cristalino eleva la energía de los orbitales metal-d-debido a la repulsión electrostática entre los electrones ligandos y los electrones metálicos.
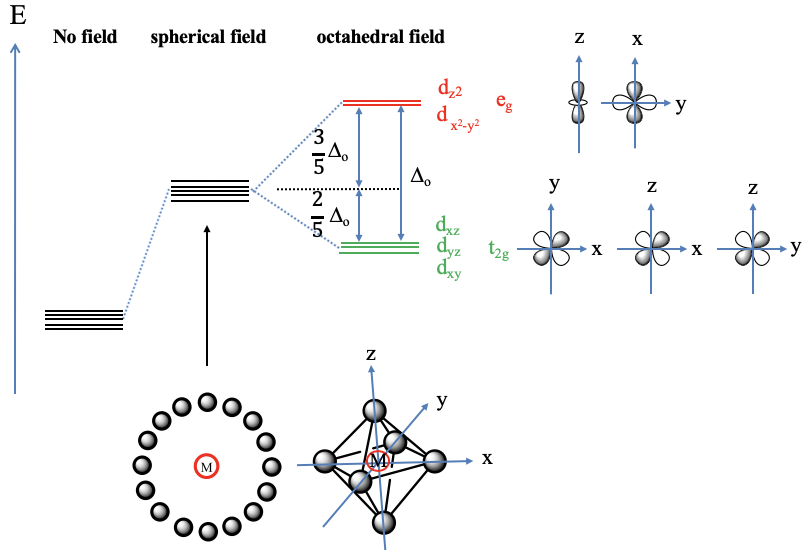
Supongamos primero el caso hipotético de que los electrones ligandos rodean los d-orbitales metálicos exactamente esféricamente (Fig. 7.1.9, abajo a la izquierda). En este caso el campo eléctrico es completamente isotrópico, y esto significa que la energía de los cinco d-orbitales metálicos aumenta en la misma medida. Ahora consideremos el caso práctico de que los ligandos rodean al metal en forma octaédrica, que es el caso en un complejo octaédrico. Podemos decir que tenemos un campo de cristal octaédrico. El campo eléctrico ya no será esférico, será el más fuerte donde estén los ligandos, es decir, en los vértices del octaedro, y menos fuerte en otros lugares. Los vértices del octaedro se encuentran en los ejes x, y y z del sistema de coordenadas. Así, el campo de cristal es el más fuerte en los ejes, y menos fuerte en otros lugares. ¿Qué consecuencia tiene esto en las energías d-orbitales metálicas en relación con el campo cristalino esférico? Los orbitales que tienen su densidad electrónica principalmente en los ejes experimentarán una mayor repulsión electrostática del campo cristalino, y por lo tanto serán más altos en energía. Los orbitales que tendrán su densidad electrónica principalmente en otros lugares, es decir, no en los ejes, experimentarán una repulsión menor, y así la energía será menor en comparación con el campo cristalino esférico. ¿Cuáles son los orbitales que tienen su densidad electrónica principalmente en los ejes? Estos son los orbitales d z 2 y d x 2 -y 2. El orbital d z 2 tiene su densidad de energía principalmente en el eje z, el orbital d x 2 -y 2 — tiene su energía principalmente en los ejes x e y. La energía de ambos orbitales se incrementa exactamente en la misma cantidad. Esto no es obvio dado que los orbitales tienen formas muy diferentes. Para entender esto, ayuda recordar que cuando los orbitales están simétricamente degenerados, también deben ser energéticamente degenerados.
Un complejo octaédrico pertenece al grupo puntual O h y en el grupo puntual O h los orbitales d z 2 y d x 2 - y 2 -orbitales son degenerados y tienen e g simetría. Por lo tanto, los orbitales d x 2 -y 2 y d z 2 -orbitales en un campo de cristal octaédrico a menudo también se denominan orbitales e g -orbitales. Los orbitales d restantes, los orbitales d xy, d yz y d xz tienen su densidad electrónica principalmente entre los ejes, por lo que su energía es menor en comparación con el campo cristalino esférico. La energía de los tres orbitales se reduce exactamente en la misma cantidad. De nuevo podemos entender esto al considerar que estos orbitales son triple-degenerados en el grupo puntual O h y tienen la simetría tipo t 2g. Por esa razón, el d xy, el d yz y el d xz en un campo cristalino octaédrico también se denominan a menudo orbitales t 2g. La diferencia de energía entre los orbitales t 2g y e g se llama Δ o. La energía de los orbitales t 2g se disminuye en 2/5 Δ o en relación con el campo cristalino esférico, y la energía de los orbitales e g se incrementa en 3/5 Δ o en relación con el campo cristalino esférico. ¿De dónde provienen los factores 2/5 y 3/5? Se deben a la ley de la conservación de la energía. La reducción global de energía debida a la disminución de energía de los orbitales t 2g -debe ser igual al aumento de energía global debido al aumento de energía de los orbitales e g: σE (t 2g) =-σ (E (e g). Debido a que hay tres orbitales t 2g pero solo dos orbitales e g esta ecuación solo se mantiene cuando la energía de los orbitales e g se incrementa en 3/5 Δ o y la energía de los orbitales t 2g disminuye en 2/5Δ o, o 3 x 2/5 Δ o = 2 x 3/5 Δ o. Obsérvese que la energía de todos los orbitales será mayor en comparación con el caso de que no exista campo eléctrico, pero la energía se incrementa en mayor medida para los orbitales e-g en comparación con los orbitales t 2g.
El campo de cristal tetraédrico
¿Y un complejo tetraédrico con un campo cristalino tetraédrico?
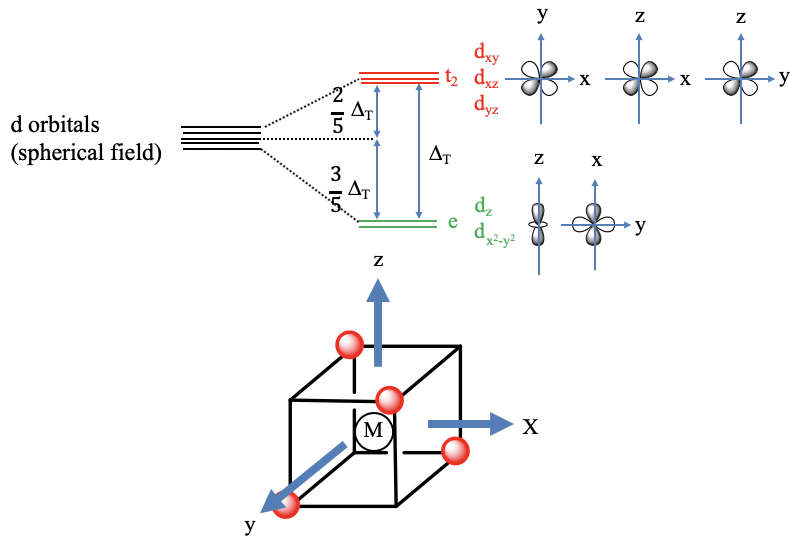
En este caso, los ligandos no se acercan sobre los ejes (Fig. 7.1.10). Esto lo podemos entender cuando consideramos que podemos inscribir un tetraedro en un cubo. Si conectamos cada otra esquina de un cubo entonces obtenemos un tetraedro. Podemos definir el sistema de coordenadas para que los tres ejes pasen por los centros de las caras cuadradas del cubo. Podemos ver que los ejes no apuntan hacia los ligandos, así los ligandos no se acercan sobre los ejes. Por lo tanto, los orbitales que tienen su densidad electrónica principalmente en los ejes tienen una energía disminuida en relación con el campo cristalino esférico. Estos son los orbitales d z 2 y d x 2 -y 2. Su energía es la misma a pesar de que tienen una forma bastante diferente. Podemos explicar esto de nuevo con argumentos de simetría. En el grupo puntual T d los orbitales d x 2 -y 2 y d z 2 — son doblemente degenerados y tienen el tipo de simetría e. Debido a que están simétricamente degenerados, también están energéticamente degenerados. La energía de los orbitales d xy, d yz y d xz tienen la mayor parte de su densidad de energía entre los ejes. Por lo tanto, sus energías se incrementan en relación con el campo cristalino esférico. Se incrementan en la misma cantidad debido a que los orbitales se degeneran triplicamente en el grupo puntual T d y tienen la simetría tipo t 2. La diferencia de energía entre los orbitales e y t 2 se llama Δ t. La energía de los orbitales t 2 se disminuye en 2/5 Δ t. La energía de los orbitales e se incrementa en 3/5 Δ t. Esto es nuevamente el debido a la ley de la conservación de la energía. La cantidad total de energía disminuida debe ser igual a la cantidad total de energía incrementada. La energía del campo cristalino tetraédrico es menor que la del campo octaédrico porque el campo octaédrico interactúa más fuertemente con los orbitales d en comparación con el campo tetraédrico. Se puede calcular que en realidad es apenas 4/9 del campo octaédrico. Esto se debe principalmente a que los lóbulos de los orbitales t 2 no apuntan exactamente hacia los vértices del tetraedro, mientras que los lóbulos de los orbitales e g sí apuntan exactamente hacia los vértices del octaedro.
Campos de Cristal Cuadrados y Octaédricos Tetragonalmente Distorsionados
Una buena característica de la teoría del campo cristalino es que puede explicar fácilmente distorsiones como la distorsión tetragonal de complejos octaédricos (Fig. 7.1.11).
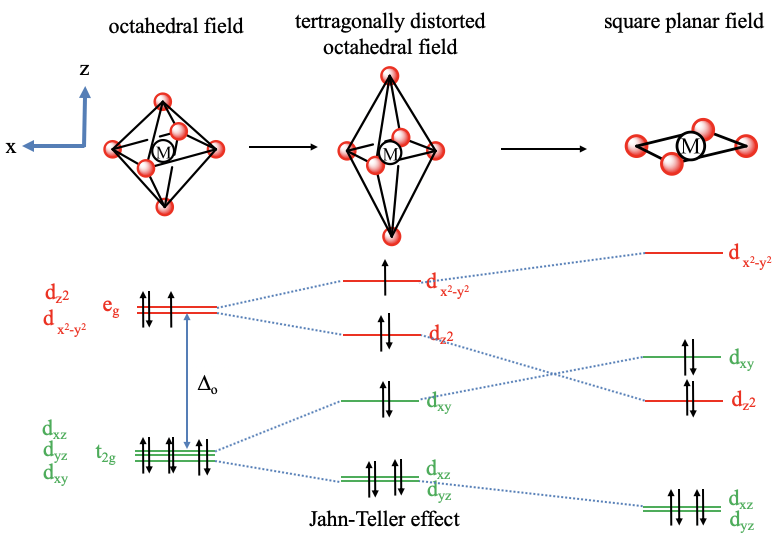
En un complejo tetragonalmente distorsionado hay un campo cristalino tetragonalmente distorsionado. En un octaedro alargado dos ligandos están más alejados del metal que los otros cuatro. Supongamos que estos dos ligandos están en el eje z. Entonces, el campo de cristal es más débil en el eje z. Para mantener constante el campo cristalino general debemos acercar los otros cuatro ligandos al centro metálico. Eso significa que comprimimos a lo largo del eje x y del eje y. ¿Qué pasará con la energía de los orbitales a medida que el octaedro se distorsione? Debido a que alargamos en la dirección z, el orbital d z 2, que tiene la mayor densidad de electrones en el eje z, baja en energía. Debido a que comprimimos en el eje x y el eje y, la energía de la órbita d x 2 -y 2 aumenta. ¿Qué pasa con los orbitales t 2g? El orbital d xy sube en energía porque tiene densidad de electrones en el plano xy, pero no a lo largo del eje z. Los d xz y d yz disminuyen en energía debido a que tienen una densidad electrónica significativa en la dirección z, y la densidad electrónica en la dirección z es la misma para ambos orbitales. Ahora también podemos pensar en, si la teoría del campo cristalino puede explicar por qué la distorsión tetragonal ocurre preferentemente para ciertas configuraciones de electrones del metal. Por ejemplo, se sabe que los iones metálicos con configuración de electrones d 9 a menudo hacen complejos octaédricos con distorsiones tetragonales. Por ejemplo, el complejo hexaaqua cobre (2+) es un ejemplo de un complejo tetragonalmente distorsionado con un ion Cu 2 + que tiene una configuración de electrones d 9. Podemos entender que la distorsión tetragonal se produce al comparar la energía de los electrones en el octaedro no distorsionado frente al distorsionado. En el octaedro no distorsionado tenemos tres electrones en los orbitales e g degenerados. A medida que nos distorsionamos, podemos mover dos electrones en el orbital energéticamente inferior d z 2 y llenar el tercero en el orbital energéticamente más alto d x 2 -y 2 -y. Así, en general los electrones tienen una menor energía explicando la distorsión. Este es un ejemplo del efecto Jahn-Teller. En general, el efecto Jahn-Teller puede ocurrir cuando hay orbitales degenerados parcialmente ocupados. En este caso una molécula puede disminuir su energía a través de la distorsión. Obsérvese que no solo la ocupación parcial de los orbitales e g, sino también la ocupación parcial de los orbitales t 2g pueden causar el efecto Jahn-Teller, aunque el efecto suele ser menor. Por ejemplo, en complejos con iones metálicos la configuración de electrones d 4, los cuatro electrones pueden estabilizarse a través de la distorsión Jahn-Teller. También hay que señalar que además de un alargamiento, también existe la posibilidad de comprimir el octaedro a lo largo del eje z. En este caso el orden de energía de los orbitales d z 2 y d x 2 -y 2 se invierte, y el orden de energía del d xy, así como el d xz y d yz se invierte también.
Finalmente, veamos el campo de cristal plano cuadrado en complejos planos cuadrados. Para entender el campo cristalino plano cuadrado ayuda a entender la forma plana cuadrada como un octaedro infinitamente alargado. Si movemos los dos ligandos a lo largo del eje z infinitamente lejos del ion metálico, entonces hemos creado una estructura plana cuadrada. ¿Cómo cambiarán las energías orbitales en comparación con un octaedro alargado? El orbital d x 2 -y 2 tendrá una energía aún mayor debido a la necesidad de compensar la disminución del campo asociado con los ligandos en el eje z comprimiendo aún más a lo largo del eje x y el eje y. El orbital d z 2 se reduce aún más en energía debido a que los ligandos a lo largo del eje z ahora han desaparecido por completo. El orbital d xy se incrementa en energía debido al campo mejorado en el plano xy. Ahora es más alto que el d z 2 -orbital. Los orbitales d xz y d yz disminuyen aún más en energía debido a que tienen una densidad electrónica significativa en la dirección z.
Ver la forma plana cuadrada como un caso extremo de un octaedro alargado tetragonalmente también nos permite entender por qué la forma plana cuadrada es adoptada tan a menudo por los complejos d 8 -metal. Podemos ver que la energía de estabilización, y así la tendencia a distorsionarse es la mayor cuando dos electrones están en los orbitales e g metálicos. En este caso dos electrones bajan su energía a través de la distorsión y ningún electrón tiene una energía incrementada. Así, entenderíamos que la distorsión se vuelve tan grande, de manera que el complejo octaédrico eventualmente pierde dos ligandos y adopta la forma plana cuadrada. Este es un buen ejemplo de cómo la teoría del campo de cristal puede explicar las formas y el número de vínculos en un complejo sin ser realmente una teoría de vinculación.
Complejos de alto giro y bajo giro
Una de las mayores fortalezas de la teoría del campo cristalino es que puede explicar los complejos octaédricos de alto y bajo espín de una manera sencilla. La base para ello es la suposición de que diferentes ligandos producen campos cristalinos de diferentes intensidades y que los campos cristalinos diferentemente fuertes producen diferentes valores Δ o. Esta suposición es plausible porque se puede esperar que diferentes ligandos interactúen de manera diferente con un ion metálico, por ejemplo, la longitud del enlace o la fuerza de unión pueden ser diferentes. Si los ligandos producen un campo cristalino grande entonces esperaríamos un δ o grande, cuando el campo cristalino es pequeño, entonces esperaríamos un δ o pequeño. El tamaño del Δ o determina si obtenemos un spin alto o un complejo de spin bajo. Si δ o es mayor que la energía de emparejamiento de espín, entonces se favorece un complejo de espín bajo, si Δ o es menor que la energía de emparejamiento de espín, entonces se favorece el complejo de espín alto.

Por ejemplo, en un complejo d 4 -metal con δ pequeño o todos los electrones están desapareados (Fig. 7.1.12, izquierda). Tres de ellos están en los orbitales t 2g, y el cuarto está en el orbital e g. Ahora supongamos un ligando diferente que puede producir un Δ o que sea lo suficientemente grande como para superar la energía de emparejamiento de espín. En este caso el cuarto electrón emparejaría el espín de uno de los tres electrones en los orbitales t 2g (Fig. 7.1.12, derecha). Obtendríamos un bajo spin d 4 -complex.
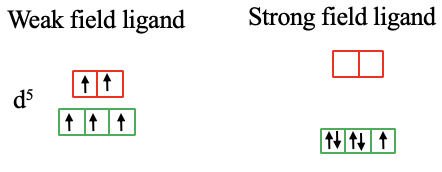
En el caso de un complejo d 5 -metal de alto espín hay cinco electrones desapareados, y todos los orbitales están medio llenos. Si el ligando produce un campo cristalino lo suficientemente grande, se supera la energía de emparejamiento de espín y hay dos electrones emparejados y un electrón desapareado en los orbitales t 2g (Fig. 7.1.13).
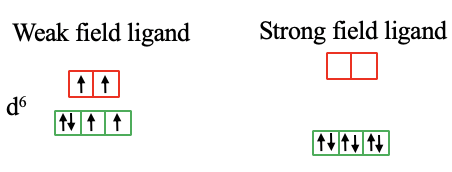
Para un complejo d 6 -metal de alto espín con un campo cristalino débil hay dos electrones desapareados en los orbitales t 2g y e g respectivamente. En el caso de un ligando de campo fuerte y un complejo de espín bajo, todos los electrones están en los orbitales t 2g y todos los espines están emparejados (Fig. 7.1.14).

En el caso de un complejo d 7 -alto espín hay dos pares de electrones y un electrón desapareado en los orbitales t 2g, y hay dos electrones desapareados en los orbitales e g. Para el complejo de bajo giro se supera la energía de emparejamiento de giros. Sin embargo, un electrón debe permanecer desapareado en los orbitales e g porque los orbitales t 2g están completamente ocupados con seis electrones. El número de electrones desapareados en complejos de espín alto y bajo predicho por la teoría del campo cristalino es lo que se observa experimentalmente. Por lo tanto, podemos afirmar que la teoría del campo cristalino puede explicar con bastante elegancia los complejos de espín alto y bajo.
También podemos entender por qué no hay d 1, d 2, d 3, d 8, d 9 y d 10 complejos de espín bajo y alto. En las configuraciones de electrones d 1 -d 3 todos los electrones están desapareados en los orbitales t 2g. Por lo tanto, no existe ningún electrón que pueda moverse de un orbital e g a un t 2g orbital. Para las configuraciones d 8 -d 10 todos los orbitales t 2g están necesariamente llenos. Por lo tanto, no hay posibilidad de mover un electrón de un orbital e g a un t 2g independientemente de la intensidad del campo cristalino.
Además, podemos explicar por qué no existen complejos tetraédricos de bajo espín. Anteriormente hemos aprendido que un campo cristalino tetraédrico tiene solo 4/9 de la fuerza de un campo octaédrico. Debido a que es mucho más débil, ningún ligando es capaz de producir un campo lo suficientemente fuerte como para superar la energía de emparejamiento de espín. Sin embargo, los complejos de alto y bajo espín son posibles para los complejos planos cuadrados.
Ligandos de campo fuertes y débiles: la serie espectroquímica
La teoría del campo cristalino no solo puede explicar bien el magnetismo, también puede hacer declaraciones sobre las propiedades ópticas de un compuesto de coordinación. Un complejo puede absorber la luz cuando un electrón es excitado de un orbital d de menor energía a un orbital d de mayor energía. Cuanto mayor sea la Δ, menor será la longitud de onda de la luz absorbida. En el caso de un complejo octaédrico, la absorción de luz excitaría un electrón de un t 2g a un orbital e g.
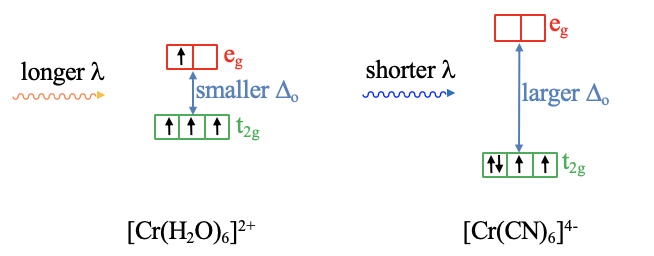
Por ejemplo, un complejo octaédrico [Cr (H 2 O)] 6 2+ tiene un Δ o menor en comparación con un complejo octaédrico [Cr (CN) 6] 4- (Fig. 7.1.16), y así el complejo aqua absorbe luz de mayor longitud de onda en comparación con el complejo ciano. Viceversa, medir el espectro de absorción de los complejos, nos permite hacer declaraciones sobre la intensidad relativa del campo cristalino de los ligandos. Al medir el espectro de absorción de muchos complejos con una variedad de ligandos podemos desarrollar una llamada serie espectroquímica que ordena ligandos de acuerdo a su intensidad de campo.
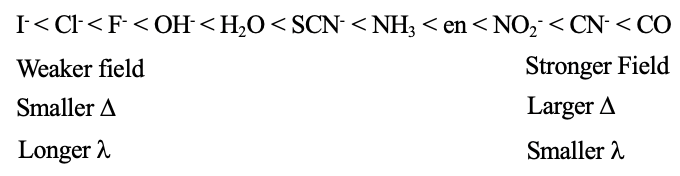
Se puede ver una serie de este tipo que contiene un número no exhaustivo de ligandos en la Fig. 7.1.17. Se puede ver que los ligandos de iodo están en el lado izquierdo, y es el ligando de campo más débil, el ligando carbonilo está en el lado derecho y es el ligando de campo más fuerte. Todos los demás están en el medio. Podemos ver por ejemplo, que un ligando aqua es un ligando de campo más fuerte en comparación con un ligando fluoro, pero un ligando más débil que un ligando de amina. Generalmente, los ligandos en el extremo inferior de la serie producen campos más débiles, Δs más pequeños y absorben la luz con longitudes de onda más largas. Los ligandos en el extremo superior de la serie producen campos más fuertes, crean Δs más grandes y absorben la luz de longitudes de onda más cortas. Sin embargo, la teoría del cristal archivado no puede explicar diferente fuerza de ligando. ¿Por qué un ligando produce un campo más fuerte que otro? Para responder a esta pregunta, necesitamos la teoría del campo ligando.
Teoría del campo del ligando
La teoría del campo de ligandos es la teoría de unión más poderosa para los compuestos de coordinación. Es esencialmente teoría orbital molecular aplicada a compuestos de coordinación. Puede explicar el enlace covalente en los compuestos de coordinación, puede explicar sus formas, puede explicar su magnetismo y sus espectros electrónicos. También puede explicar la estabilidad de los compuestos de coordinación, en particular la regla 18e y sus excepciones. Es, sin embargo, más complicado que otras teorías de vinculación.
Al igual que en la teoría de orbitales moleculares, podemos aplicar una combinación lineal adaptada a simetría de orbitales atómicos para construir orbitales moleculares. Sin embargo, se aplican reglas ligeramente modificadas para optimizar la teoría orbital molecular para compuestos de coordinación. Estas modificaciones son útiles debido a la mayor complejidad de los compuestos de coordinación. En el primer paso determinamos el grupo puntual de la molécula y asignamos ejes de manera útil. En el segundo paso, determinamos los orbitales de valencia, también llamados orbitales fronterizos del metal. Por ejemplo, para un metal de transición del 4to periodo consideraríamos los orbitales 4s, 4p y 3d como orbitales fronterizos. A continuación, determinamos la simetría de estos orbitales. Podemos hacer esto con sólo mirar en la tabla de caracteres del grupo de puntos respectivo. A continuación seleccionamos los orbitales de ligando de mayor energía que son adecuados para el enlace σ-bonding. Para ligandos que son moléculas o iones poliatómicos, estos orbitales son los HOMO adecuados para la unión σ-enlace. Para los iones simples como los cloro-ligandos, estos son los orbitales atómicos más altos ocupados. A continuación, agrupamos los orbitales de ligando seleccionados para formar orbitales de grupos de ligandos (LGO) y determinar sus tipos de simetría. Para ello, determinamos la representación reducible, y luego las representaciones irreducibles de las OGs. Esto nos da los tipos de simetría de los orbitales del grupo ligando. Luego combinamos orbitales de frontera metálica y orbitales de grupos de ligandos del mismo tipo de simetría para formar orbitales moleculares. Ahora hemos construido todos los orbitales moleculares adecuados para la unión σ, y podemos dibujar un diagrama orbital molecular para el enlace σ-en el compuesto de coordinación.
A continuación, buscamos orbitales de ligandos que sean adecuados para la unión π con el metal. Luego determinamos los tipos de simetría de sus orbitales de grupos de ligandos. Luego se combinarán orbitales de grupos de ligandos y orbitales metálicos de la misma simetría para formar orbitales moleculares. Estos MO representan el enlace π en la molécula. Agregamos estos orbitales moleculares al diagrama orbital molecular. Finalmente, verificamos si hay orbitales de ligandos adecuados para la\(\delta\) unión con el metal. Si es así, también formamos orbitales de grupos de ligandos para estos, determinamos sus tipos de simetría y combinamos orbitales de grupos de ligandos y orbitales metálicos de la misma simetría para formar orbitales moleculares. Estos orbitales se añadirán entonces también al diagrama orbital molecular.
Reglas aplicables para la construcción de diagramas orbitales moleculares usando teoría de campo de ligandos
- Determinar el grupo de puntos del complejo y asignar ejes.
- Determinar cuáles son los orbitales de frontera metálica relevantes para la unión.
- Determinar los tipos de simetría de estos orbitales metálicos.
- Seleccione los homOS del ligando (adecuados para la unión a) si el ligando es una molécula. Si es un ion simple, seleccione el orbital atómico ocupado más alto.
- Formar orbitales de grupos de ligandos (LGO) a partir de orbitales de ligandos seleccionados y determinar sus tipos de simetría.
- Combinar orbitales metálicos y orbitales de grupos de ligandos de simetrías apropiadas para formar orbitales moleculares.
- Seleccionar orbitales de ligando\(π\) y\(σ\) unirlos si corresponde, determinar su simetría y combinarlos con orbitales metálicos apropiados.
Teoría del campo de ligando para un complejo octaédrico de un metal de transición del 4to periodo
Apliquemos primero la teoría del campo ligando a un complejo octaédrico de metal de transición del 4to período. De acuerdo con las reglas primero debemos determinar el grupo de puntos y definir el sistema de coordenadas. El grupo de puntos es obviamente O h.
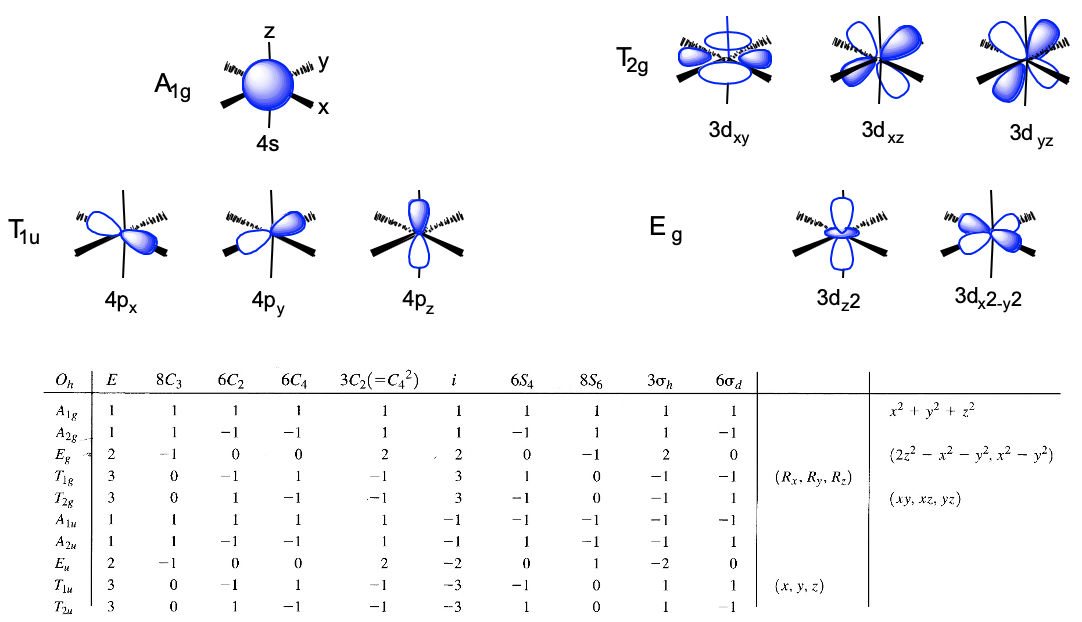
Podemos definir el sistema de coordenadas para que los ligandos se acerquen en los ejes x, y y z, respectivamente. A continuación, necesitamos ver cuáles son los orbitales fronterizos. Estos serían los orbitales 4s, 4p y 3d (Fig. 7.1.18). ¿Qué tipos de simetría tienen? Esto lo podemos ver buscando en la tabla de caracteres para el grupo de puntos O h. Un orbital s siempre tiene el tipo de simetría totalmente simétrica que siempre aparece primero en la tabla de caracteres. En el grupo de puntos O h esta es la simetría tipo A 1g. ¿Qué pasa con los orbitales 4p? Podemos ver que las letras x, y y z están entre paréntesis en la representación irreducible del tipo de simetría T 1u. Esto significa que los tres orbitales 4p son triplicamente degenerados y tienen el tipo de simetría T 1u. Finalmente, necesitamos determinar los tipos de simetría de los orbitales 3d. Encontramos xy, xz e yz entre paréntesis en la representación irreducible del tipo de simetría T 2g. Así, estos orbitales tienen la simetría tipo T 2g. Encontramos además 2z 2 -x 2 -y 2 y x 2 -y 2 en la línea para el tipo de simetría E g, así los orbitales d z 2 y d x 2 -y 2 son degenerados y tienen el tipo de simetría E g. Recuerda aquí del capítulo sobre la estructura atómica que 2z 2 -x 2 -y 2 describe matemáticamente un cono, y que el orbital d z 2 tiene un nodo cónico. Ahora hemos encontrado todos los tipos de simetría de los orbitales fronterizos.
A continuación, necesitamos dirigir nuestra atención hacia el ligando y encontrar el orbital molecular u atómico más alto ocupado adecuado para la unión σ-bonding. Por supuesto, depende ahora cuál es el ligando.
Como ejemplo escojamos el ligando carbonilo común e interesante (Fig. 7.1.19). Para determinar su HOMO adecuado para la unión σ con el metal, primero necesitaremos conocer el diagrama orbital molecular de la molécula de monóxido de carbono. La molécula de monóxido de carbono es una molécula lineal, polar que pertenece al grupo puntual C ∞ v.

La tabla de caracteres de este grupo de puntos es algo difícil de trabajar debido al orden infinito del eje principal, y al número infinito de planos especulares verticales. Por lo tanto, usaremos en su lugar el subgrupo C 4v (Fig. 7.1.20). Un subgrupo de un grupo es un grupo que resulta cuando eliminamos ciertos elementos de simetría del grupo original. Deberíamos eliminar los elementos de simetría para que no se pasen por alto las degeneraciones en los orbitales moleculares, lo que puede suceder cuando se reduce demasiado la simetría. El grupo de puntos C 4v es el grupo de puntos con la simetría más baja que podemos elegir sin pasar por alto las degeneraciones. Esencialmente, esto se debe a que los orbitales atómicos de C y O son solo 2s y 2p, y los orbitales 2p perpendiculares al eje de enlace C-O pueden girarse 90° para hacer que los orbitales se interconviertan. Esto requiere un eje de rotación del orden 4. Si elegimos el grupo puntual C 2v, que tiene simetría aún menor, aún podríamos construir un diagrama orbital molecular, pero pasaríamos por alto que los orbitales 2p x y 2p y están degenerados. Vemos que en el caso de una molécula lineal diatómica no hay átomo central ni ligandos. Por lo tanto, no podemos aplicar el método SALC exactamente como lo aprendimos anteriormente. Por lo tanto, tratamos a los átomos de C y O como orbitales de átomos centrales que interactúan entre sí. Para determinar sus tipos de simetría solo podemos mirar en la tabla de caracteres para C 4v. Encontramos que los orbitales 2s y los orbitales 2p z tienen la simetría tipo A 1 y los orbitales 2p x y 2p y son doblemente degenerados y tienen la simetría tipo E (Fig. 7.1.20). Ahora sólo podemos combinar los orbitales atómicos para formar orbitales moleculares.
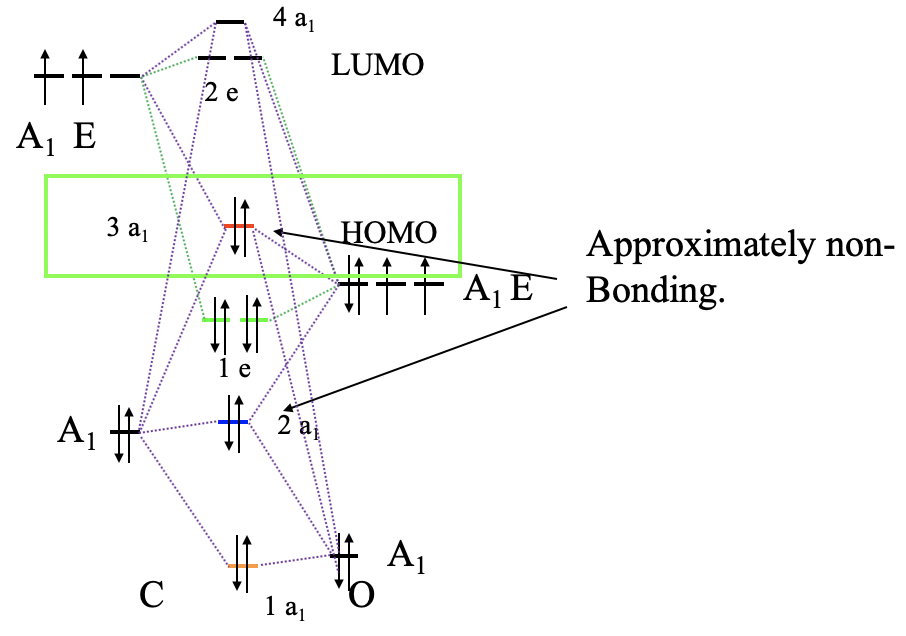
Para construir un diagrama orbital molecular debemos considerar que O es considerablemente más electronegativo que C, y por lo tanto, el orbital 2s de O es menor en energía en comparación con el orbital 2s de C. Los orbitales 2p de O son más bajos en energía que los de C. Ahora podemos etiquetar nuestros orbitales con sus tipos de simetría, y combinar orbitales del mismo tipo de simetría para formar orbitales moleculares (Fig. 7.1.21). Podemos comenzar, por ejemplo, con los orbitales del tipo de simetría A1. Hay dos en cada lado, así que tendremos en general cuatro. Nuestra suposición cualitativa sería que debería haber una fuerte unión y un orbital fuertemente antienlace, y débilmente enlace y un orbital débilmente antienlace además. El que se une fuertemente tendría la energía más baja y estaría etiquetado como 1a 1, el que se une débilmente tendría mayor energía y está etiquetado como 2a 1, el débilmente antiunión tiene una energía aún mayor y está etiquetado como 3a 1, y el fuertemente antienlace tendría el más alto energía de los cuatro, y tiene la etiqueta 4a 1. El orbital fuertemente antienlace debe estar por encima del orbital 2p z de C, y el fuertemente ligado debe estar por debajo del orbital 2s de O. Podemos escribir el orbital 2a 1 y el 3a 1 a niveles de energía para que las diferencias de energía entre los cuatro orbitales a 1 sean más o menos lo mismo. Ahora también podemos conectar los MO y los AO de una simetría 1 con líneas punteadas. Ahora podemos dirigir nuestra atención a los orbitales con simetría E. En general, hay cuatro orbitales de simetría E, por lo tanto, esperamos dos orbitales vinculantes dobles—degenerados con e-simetría, y dos orbitales anti-enlace doblemente degenerados con e-simetría. Los MO de unión deben estar por debajo del nivel de energía de los orbitales 2p de O, y los dos orbitales antiunión deben escribirse por encima de los niveles de energía de los orbitales 2p de C. Ahora podemos conectar los orbitales atómicos y moleculares con líneas punteadas. Por último, todavía tenemos que rellenar los electrones. Hay cuatro electrones que vienen del carbono y seis electrones que vienen del oxígeno, lo que da diez electrones en general. Esto significa que los orbitales moleculares 1a 1, 2a 1, 1e 1 y 3a 1 están llenos. Esto hace que el 3a 1 orbital sea el HOMO. Es adecuado para la unión σcon el metal porque se ha creado a través de interacciones σentre los orbitales 2p z y 2s de O y P respectivamente.
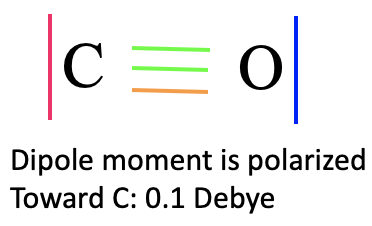
De nuevo es interesante comparar la imagen MO de la unión en CO con la estructura Lewis-dot (Fig. 7.1.19). En la estructura de puntos de Lewis tenemos un triple enlace con seis electrones de unión. Corresponden a los electrones de unión 1a 1 y 1e. En la estructura de puntos de Lewis los seis electrones son energéticamente indistinguibles, pero en el diagrama MO podemos ver claramente que dos electrones tienen una energía menor que los otros cuatro que están energéticamente degenerados. El 1a 1 MO es un σ-orbital mientras que los otros dos son π-orbitales debido a que los orbitales 2p x y 2p y interactúan de manera π-moda. Los orbitales 2a 1 y 3a 1 pueden aproximarse como MO no enlazantes que representan los dos pares de electrones solitarios en C y O respectivamente. Debido a que el 3a 1 es ligeramente antienlace tiene su densidad electrónica principalmente en C, mientras que el 2a 1 se une ligeramente y por lo tanto su contraparte es el par solitario de electrones en O. Curiosamente el momento dipolar del CO está ligeramente polarizado hacia C por 0.1 Debye a pesar de la hecho de que O es el átomo más electronegativo. Esto se puede atribuir a que el HOMO como orbital débilmente antienlace se ubica principalmente en C, Además, el orbital 2a 1 está bastante cerca en energía a los 2s de C, por lo tanto los 2s de C contribuyen significativamente a este orbital. Esto lleva al hecho de que también hay una cantidad significativa de densidad de electrones localizada en C.

En el siguiente paso, necesitamos agrupar los seis HOM para formar orbitales de grupos de ligandos y determinar sus tipos de simetría. Esto se hace determinando primero la representación reducible de los orbitales utilizando el método de intercambio orbital, y luego determinando el número de representaciones irreducibles de un tipo dado usando la fórmula de reducción de la teoría de grupos. Aquí no vamos a pasar por los detalles de los cálculos, porque no hay nada realmente nuevo que aprender, sino simplemente discutir el resultado. El resultado es que hay un grupo ligando orbital con simetría tipo a 1g, dos orbitales de grupo ligando doblemente degenerados con el tipo de simetría e g, y tres orbitales de grupo ligando triplicamente degenerados que tienen el tipo de simetría t 1u. El orbital a 1g no tiene un nodo porque es totalmente simétrico. Los orbitales t 1u tienen un nodo, y los orbitales e-g tienen dos nodos (Fig. 7.1.22). Esto no es resultado de la fórmula de reducción, pero se podría demostrar que calculando realmente los orbitales del grupo de ligandos usando una fórmula llamada operador de proyección (que no discutiremos en detalle aquí).
Debido a que ahora conocemos nuestros tipos de simetría de nuestros orbitales de frontera metálica y nuestros orbitales de grupos de ligandos, podemos construir un diagrama orbital molecular cualitativo (Fig. 7.1.23). Para ello podemos trazar los orbitales fronterizos en el lado izquierdo del diagrama orbital molecular, y los orbitales del grupo ligando en el lado derecho del diagrama orbital molecular.
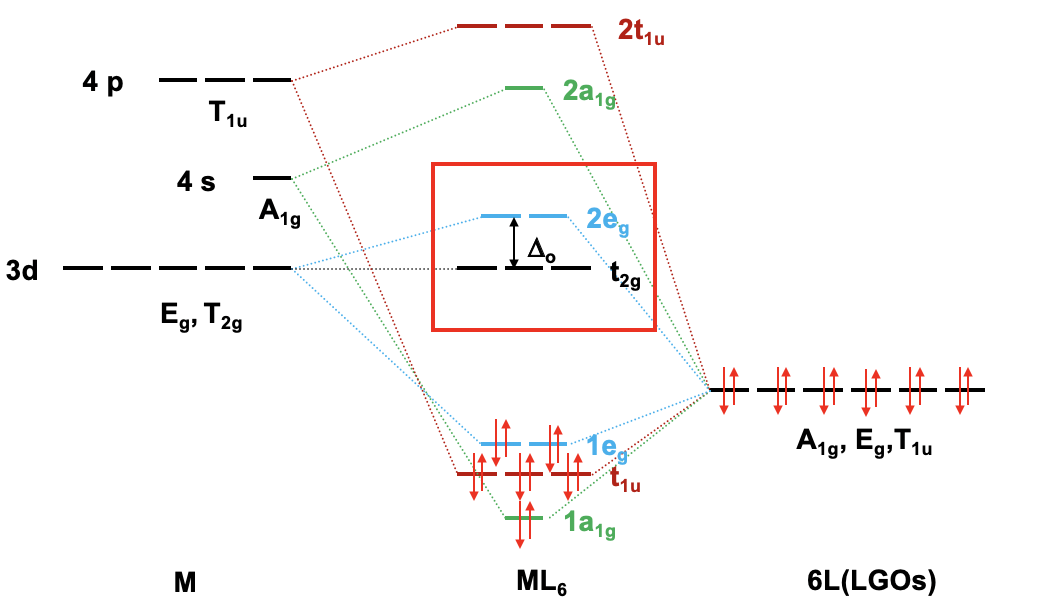
Para un metal de transición de 4ª fila la secuencia de energía es 3d<4s<4p. Tiene sentido suponer que los orbitales del grupo ligando tienen aproximadamente la misma energía que los orbitales 3d del metal, y podemos trazarlos en consecuencia. A continuación, podemos asignar a los orbitales sus tipos de simetría previamente determinados. Entonces, podemos comenzar a combinar orbitales de los mismos tipos de simetría para formar orbitales moleculares. Podemos comenzar por ejemplo con los orbitales que tienen la simetría tipo A1. El orbital 4s tiene ese tipo de simetría. También un grupo ligando orbital es de este tipo. Por lo tanto, esperaríamos un orbital molecular de enlace y uno antienlace. Podemos etiquetarlos 1a 1g y 2a 1g, respectivamente, y conectarlos con los orbitales del grupo atómico y ligando A 1g mediante líneas punteadas. A continuación, por ejemplo, podemos considerar los orbitales E g. Hay dos orbitales de metal d y dos orbitales de grupos de ligandos de esa simetría. Por lo tanto, producimos dos enlaces doblemente degenerados y dos orbitales moleculares anti-enlace doblemente degenerados. Podemos etiquetarlos 1e g y 2e g respectivamente. Podemos ver además, que existen los tres orbitales T 1u metal 4p que podemos combinar con los orbitales T 1u del ligando. Esto da tres orbitales de unión triple degenerado y tres orbitales antiunión triple degenerados con simetría t 1u. Por último, están los orbitales metálicos T 2g. No hay orbitales de grupos de ligandos con la misma simetría, y por lo tanto los orbitales T 2g permanecen sin unión. Tenemos que escribirlos con energía inalterada en el diagrama orbital molecular.
Ahora hemos terminado con la construcción de orbitales moleculares, pero aún necesitamos llenar los electrones en ellos. Consideramos que el enlace metal-ligando es un enlace dativo, siendo los pares de electrones donados por el HOMO del ligando. Por lo tanto, consideramos que todos los orbitales del grupo de ligandos están llenos de electrones. Eso significa que tenemos en general 6x2=12 electrones a considerar. ¿Qué pasa con los electrones metálicos? Bueno, ahora depende de qué ion metálico tengamos. Supongamos aquí que tenemos un ión metálico d 0 sin electrones d. Eso significa que en general tenemos 12 electrones que necesitamos llenar en los orbitales moleculares según la energía. Esto llena los orbitales 1a 1g, tres t 1u, y los dos 1e g. Ahora supongamos que no tendríamos un ión metálico d 0, sino un ion metálico con d electrones. Podría haber hasta diez d electrones. ¿A dónde irían? Entrarían a los cinco orbitales con las siguientes energías más altas. ¿Cuáles son estos? Bueno, estos son los orbitales t 2g y 2e g. Los orbitales t 2g son los orbitales de metal no enlazante d xz, d xy y d yz, por lo que estos orbitales se localizan solo en el metal. Los orbitales 2e g son orbitales moleculares antienlace que se han construido a partir de los orbitales d z 2 y d x 2 -y 2, y son similares en energía a los d x 2 -y 2 y d z 2 orbitales. Por lo tanto, podemos decir que los orbitales t 2g y 2e g son los orbitales d bajo la influencia de un campo ligando octaédrico. Debido a la presencia del campo ligando las energías de los d-orbitales metálicos se dividen y la diferencia en energía es la energía octaédrica del campo ligando\(\Delta\) o. Ahora podemos ver que existe una interesante analogía con la teoría del campo cristalino. Al igual que los orbitales d se dividieron en energía bajo un campo de cristal octaédrico en orbitales t 2g y e g, los orbitales d se dividieron en energía en un campo de ligando octaédrico en orbitales t 2g y e g. Al igual que en la teoría del campo cristalino se eleva la energía del d z 2 y del d x 2 -y 2. La energía de los otros tres orbitales d no se ve afectada similar, pero no exactamente la misma que los orbitales d xy, d yz y d xz en un campo cristalino octaédrico.
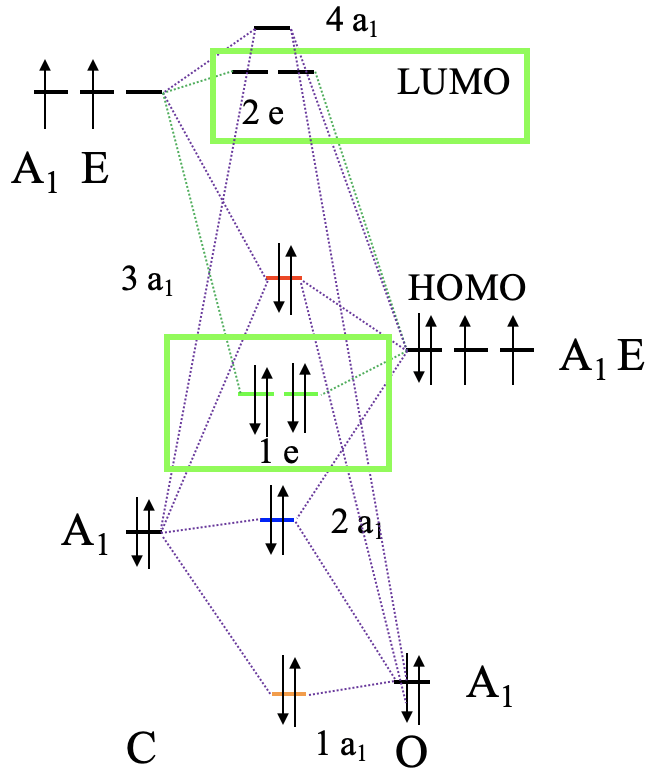
Ahora hemos entendido el enlace σen un complejo octaédrico, a continuación consideremos el enlace π. Eso significa que tenemos que pensar en qué orbitales del ligando tiene la simetría correcta para la unión π con el metal. También deben tener una energía similar a los orbitales metálicos para que pueda ocurrir una interacción covalente significativa. Nos quedaremos con nuestro ligando carbonilo, por lo que necesitamos investigar nuevamente el diagrama orbital molecular de la molécula de CO, y ver si hay orbitales moleculares adecuados para el enlace π (Fig. 7.1.24). La molécula de CO tiene los orbitales 1e y 2e, que son orbitales π-orbitales de enlace y antienlace, respectivamente. Podemos entender esto al considerar que están construidos a partir de los orbitales 2p x y 2p y que se superponen en forma π-para hacer el enlace π en la molécula. Cada ligando tiene dos orbitales 1e y dos 2e, lo que da cuatro orbitales en general. Estos orbitales son energéticamente similares al HOMO, por lo que podemos esperar que estén lo suficientemente cerca energéticamente a la energía de los orbitales d-metal, y puedan involucrarse en la vinculación. Tenemos seis ligandos, lo que significa que tenemos 6x12=24 orbitales en general. Los doce orbitales 1e son orbitales π-enlazantes, y los doce orbitales 2e son orbitales π*-antienlace.
¿Por qué estos orbitales son adecuados para la unión π con orbitales d metálicos? Veamos su forma, y cómo pueden superponerse con un d-orbital metálico (7.1.25).
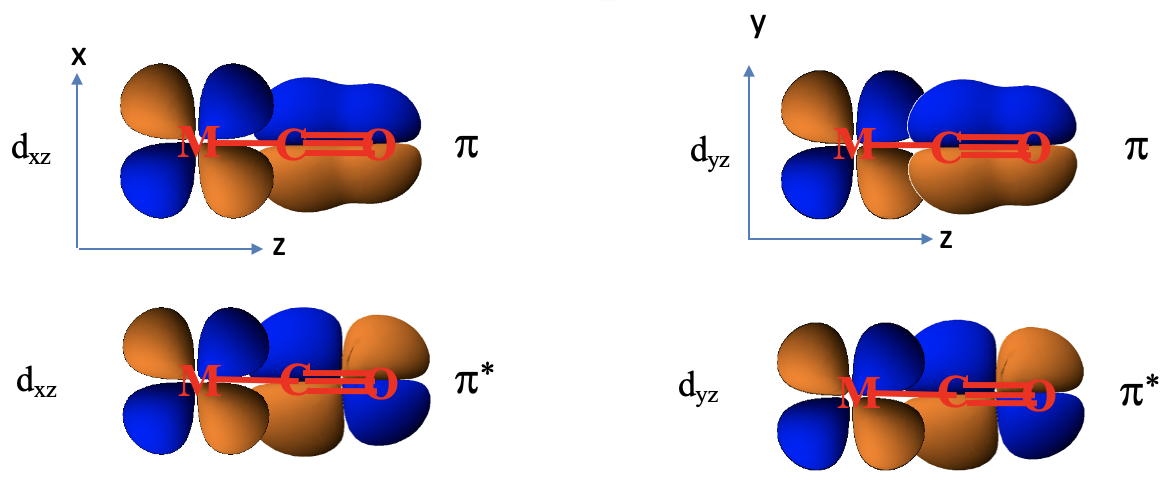
Mire por ejemplo un orbital 1e construido a partir de dos orbitales 2p x, y cómo se orienta en relación con el eje de enlace metal-ligando que definiremos como el eje z. A continuación, consideremos un metal d xz orbital orbital. Podemos ver que está en plano con el orbital 1e, y que el solapamiento entre el orbital d xz y el 1e ocurre de manera π-fashion. Ahora consideremos el análogo π*-orbital. Podemos ver que tiene un nodo adicional, pero también puede superponerse con un orbital d xz en forma π-fashion. El ligando no solo tiene orbitales π y π*-mediante superposición de dos orbitales 2p x, sino también orbitales π y π*-que resultan del solapamiento de dos orbitales 2p y. Esos orbitales pueden superponerse en forma π-con un orbital de metal d yz. Hasta el momento, se consideraron las interacciones de un solo ligando con el metal solamente. Hay otros cinco ligandos, uno también acercándose desde el eje z, y los otros cuatro acercándose desde los ejes x e y. También tienen los orbitales π y π*-que interactúan con los orbitales d xz, d yz o d xy en forma π, dependiendo de la dirección de aproximación.
Entonces, ¿qué hacemos con todos estos orbitales? Agrupamos los doce enlaces para formar un conjunto de orbitales del grupo ligando y los doce antienlaces para formar otro conjunto de orbitales del grupo ligando. Determinamos los tipos de simetría de cada conjunto determinando primero la representación reducible, y luego las representaciones irreducibles usando la fórmula de reducción. El resultado de este proceso es que los doce orbitales del grupo ligando de unión tienen simetría T 1g, T 2g, T 1u y T 2u. Esto quiere decir que hay tres triple-degenerados que tienen simetría T 1g, otros tres triple-degenerados que tienen T 1u, simetría, tres más triple-degenerados que tienen simetría T 1u, y otros tres triple-degenerados que tienen T 2u simetría. Los doce orbitales del grupo de ligandos antiunión tienen los mismos tipos de simetría. Tres tienen simetría T 1g, tres tienen simetría T 2g, tres tienen simetría T 1u y otros tres tienen simetría T 2u.
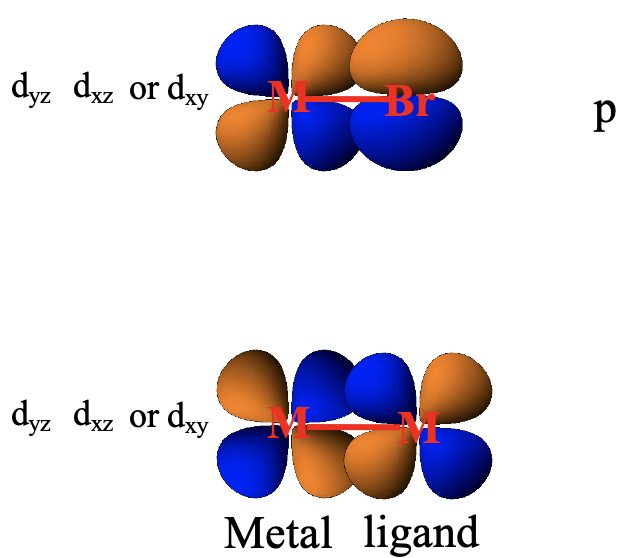
Cabe mencionar que hay una serie de otros orbitales que pueden hacer interacciones π-cuando el ligando no es un ligando CO. Por ejemplo, los ligandos de halógeno tienen orbitales p que tienen una orientación adecuada para superponerse con orbitales d metálicos en forma π (Fig. 7.1.26). Cuando hay complejos con enlaces metal-metal, entonces también existe la posibilidad de que los orbitales d metálicos se superpongan con otros d-orbitales metálicos en forma π.
Ahora que hemos determinado los tipos de simetría de los orbitales del grupo ligando disponibles para la unión p, necesitamos seleccionar aquellos orbitales de grupos de ligandos que tengan simetría adecuada para formar orbitales moleculares con orbitales d metálicos en el campo de ligandos octaédricos. Estos son los 2e g y los t 2g MO que resultaron de las interacciones σ. Por simplicidad, llamaremos a los orbitales moleculares 2e g solo los orbitales metálicos e g. Los t 2g MO son idénticos a los orbitales atómicos T 2g metálicos porque los orbitales atómicos T 2g metálicos permanecen exactamente sin unión con respecto a las interacciones σ-. Los orbitales del grupo ligando tienen simetría T 1g, T 2g, T 1u y T 2u respectivamente. Eso significa que podemos combinar los orbitales metálicos t 2g y los orbitales del grupo ligando T 2g para formar orbitales moleculares. Los orbitales e g metálicos y los orbitales del ligando T 1g, T 1u y T 2u permanecen sin unión porque no encuentran pareja (Fig. 7.1.27). Los tres T 2g LGO de unión formarán seis MO del mismo tipo de simetría con los tres orbitales metálicos t 2g. Además, hay que considerar que también hay tres T 2g * LGO antiadherentes que se formaron a partir de los orbitales π-enlaces antiadherentes. También pueden interactuar con los orbitales d de metal t 2g. Debido a que el número de MO de un tipo de simetría dado es siempre la suma de los orbitales atómicos + los LGO de ese tipo de simetría esto agrega tres MO a los seis MO dando en general nueve t 2g MOs. También están los orbitales T 1u * , T 1g * y T 2u *. Simplemente permanecen sin vinculación porque no encuentran pareja.
Ahora podemos pensar en dos casos extremos para la combinación del metal t 2g con los orbitales ligando T 2g y T 2g *. En el primer caso los T 2g LGO son mucho más cercanos en energía a los orbitales metálicos t 2g, y los T 2g * LGO son energéticamente mucho más altos que los orbitales metálicos t 2g. En este caso podemos descuidar las interacciones covalentes entre los orbitales T 2g * y los orbitales metálicos t 2g, y los orbitales T 2g * permanecen efectivamente sin unión. Consideraríamos solo las interacciones entre los T 2g LGO y los orbitales metálicos t 2g para formar tres MO de unión y tres anti-unión de simetría t 2g (Fig. 7.1.28). Ahora tenemos que considerar los electrones. Los T 2g LGO de unión están llenos, y por lo tanto hay en general seis electrones a considerar. Estos seis electrones entrarían en los tres mOS de t 2g de unión. Ahora también puede tener hasta diez electrones d metálicos. Seis de ellos pueden ser acomodados por los orbitales metálicos t 2g, los otros cuatro estarían en los orbitales e g. Tras la formación del enlace π, los electrones metálicos t 2g estarán en los orbitales anti-unión t 2g. Los electrones e g simplemente permanecerán sin unión. Podemos ver que las interacciones π-disminuyen la energía de los electrones ligandos, pero aumentan la energía de los electrones metálicos. Si conseguimos una estabilización neta de las energías de electrones dependerá de cuántos d electrones tengamos. Mientras haya menos de seis d electrones veremos una estabilización, si hay más habrá una desestabilización general. También podemos preguntar qué impacto tiene la unión π-en la magnitud del Δ o. Podemos ver que el enlace π disminuye el Δ o. Es más grande antes de la unión en comparación con después de la unión π.
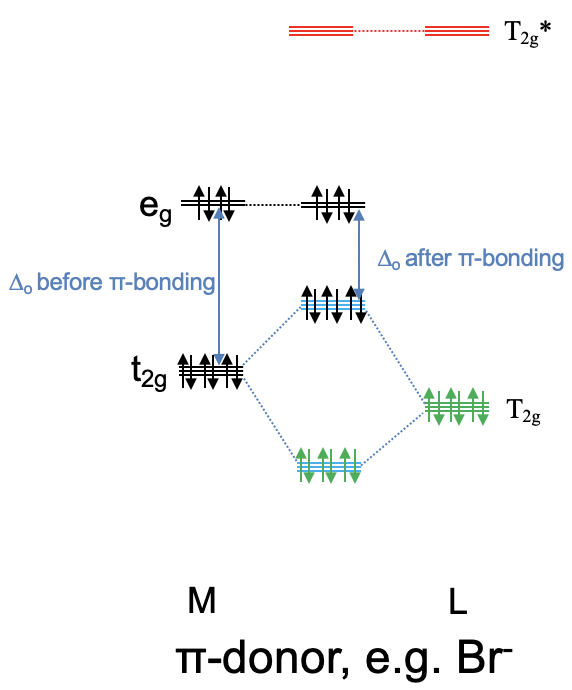
Llamamos a un ligando que tiene orbitales T 2g de energía similar al metal t 2g orbitales, y T 2g * orbitales de energía mucho mayor en comparación con los orbitales metálicos t 2g un ligando donador de π, o un donador π (Fig. 7.1.28). Esto se debe a que antes del enlace p los electrones del ligando se localizan exclusivamente en el ligando, y después del enlace p están en el enlace t 2g MOS que se comparten entre el metal y el ligando. Así, se ha producido una transferencia parcial de electrones del ligando al metal. Un ejemplo de ligandos donadores de p son ligandos de halogenuro tales como un ligando de bromo.
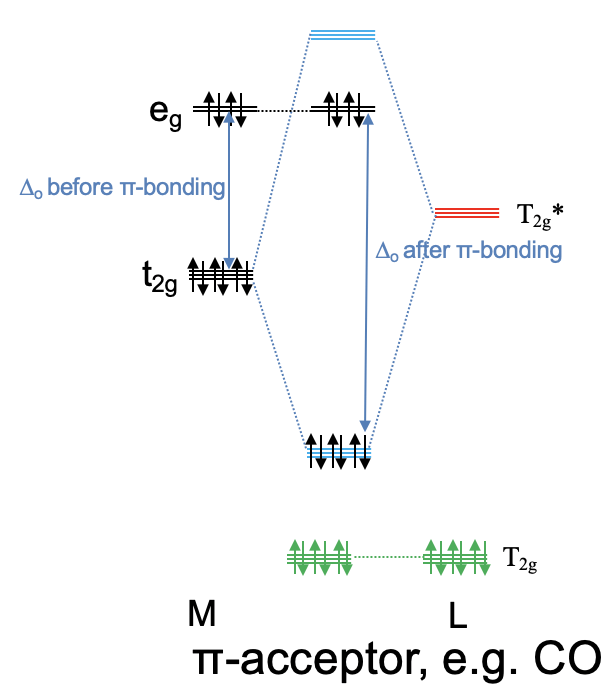
Ahora consideremos el caso opuesto en el que los orbitales del ligando T 2g * antiunión están energéticamente cerca de los orbitales metálicos t 2g y los orbitales del ligando T 2g de unión son energéticamente demasiado bajos para interactuar significativamente con el metal t 2g orbitales. Esto significa que los orbitales T 2g permanecen prácticamente sin unión. Los orbitales del grupo ligando T 2g * y los orbitales metálicos t 2g forman tres orbitales moleculares de enlace triple degenerado y tres orbitales moleculares antienlace triples degenerados. Debido a que los orbitales del grupo ligando T 2g * están vacíos, no se llenan electrones ligandos en los nuevos orbitales moleculares. Esto significa que se pueden llenar hasta seis electrones d metálicos en los orbitales moleculares de unión de simetría t 2g. Todos los electrones d restantes en los orbitales e g simplemente permanecen en los orbitales e g y no cambian de energía. Podemos ver que en contraste con el caso anterior, podemos disminuir la energía de los electrones del metal d a través de interacciones π-. Debido a que los electrones de unión t 2g se comparten entre el metal y el ligando, la densidad de electrones se ha transferido del metal al ligando. Por lo tanto, un ligando que utiliza principalmente sus orbitales anti-unión T 2g * para la unión π se llama ligando aceptor π, acepta densidad de electrones d del metal. Un ejemplo para un ligando aceptor p es el ligando carbonilo (Fig. 7.1.29). También es importante entender la influencia de un ligando aceptor π en el tamaño de Δ o. Debido a que el t 2g MO antiunión es mayor en energía que los orbitales e g no enlazantes, el Δ o ahora se define por la diferencia de energía entre los t 2g MOs de unión y los orbitales e g (Fig. 7.1.29). Podemos ver que Δ o se incrementa cuando se toman en cuenta las interacciones π-aceptor.
Ahora hemos discutido los dos casos extremos, sin embargo hay muchos ligandos que en realidad están entre estos dos extremos, y hay un espectro continuo de ligandos fuertemente donadores de π, débilmente donadores de π, débilmente aceptores de π, a ligandos fuertemente aceptores de π. También es posible que los efectos donadores de π y aceptores de π se cancelen. Este es el caso cuando los orbitales del grupo T 2g y T 2g * -ligando son energéticamente aproximadamente equidistantes a los orbitales metálicos t 2g. Algunos ligandos tampoco tienen orbitales adecuados para el enlace π en absoluto, y no hay efectos donadores ni aceptores de π.
El efecto de la unión π-en Δ o puede explicar muy bien la serie espectroquímica. Debido a que los ligandos aceptores de π aumentan Δ o, estos complejos absorben luz de longitud de onda más corta y de mayor energía. Los ligandos donadores de π disminuyen Δ o y por lo tanto conducen a la absorción de luz que tiene longitudes de onda más largas. Vemos aquí que la teoría del campo de ligandos es más poderosa que la teoría del campo cristalino. Este último no pudo explicar por qué diferentes ligandos producen diferentes valores Δ o.
La teoría del campo de ligandos también es capaz de explicar muy bien el magnetismo de los compuestos de coordinación, y los complejos de espín alto y bajo en particular. También es capaz de explicar por qué ciertos ligandos tienden a producir complejos de bajo espín, mientras que otros tienden a formar complejos de alto espín. Según la teoría del campo de ligandos, los aceptores π-tienden a hacer complejos de espín bajo, y los donadores π tienden a hacer complejos de espín alto. Esto está de acuerdo con la observación experimental.
Complejos octaédricos y la regla de los 18 electrones
Figura 7.1.30 Diagrama MO cualitativo del complejo octaédrico hexacarbonil-cromo bajo consideración de\(\pi\) unión.
Otra gran característica de la teoría del campo de ligandos es que puede explicar la regla de 18 electrones, y las excepciones de la regla de 18 electrones. Por ejemplo, el complejo octaédrico hexacarbonilcromo es un complejo de 18 electrones. Vamos a construir un diagrama orbital molecular cualitativo y ver si el diagrama MO soporta la estabilidad del complejo. El diagrama MO considerando solo las interacciones σse muestra en la Fig. 7.1.30. Podemos ver que todos los electrones del ligando twlelve están en los orbitales moleculares de unión 1a 1, 1t u y 1e g. Además el cromo tiene seis electrones de valencia. Estos electrones permanecen sin unión cuando se consideran interacciones σ-solamente. Sin embargo, esto cambia, cuando consideramos las interacciones π. El ligando CO es un ligando aceptor p fuerte, por lo que consideramos solo orbitales de su grupo T 2g * π-ligando para la unión. Deben estar ubicados energéticamente por encima de los LGO para la unión σ. La interacción de los orbitales metálicos t 2g crea tres mOS de unión t 2g y los tres antiadherentes. Debido a que podemos llenar electrones de metal d en los MOS de unión, el estado de los electrones d ha cambiado de no unión a unión. Podemos ver ahora que todos los 18 electrones están en MO de unión, y que ningún electrón es MO no enlazante o anti-unión. Cuando todos los electrones están uniendo MO entonces esta es la situación ideal para una estabilidad compleja. Esto explica por qué el complejo de 18 electrones es estable. Si analizamos los diagramas MO de muchos otros complejos estables de 18 electrones entonces también encontraríamos principalmente que todos los MO de unión están llenos, y todos los demás MO están vacíos. Esto explica la regla de los 18 electrones.
Figura 7.1.31 El diagrama orbital molecular cualitativo de WCl 6 bajo consideración\(\pi\) de unión.
A continuación, construyamos un diagrama orbital molecular cualitativo de WCl 6. Este no es un complejo de 18 electrones, solo tiene doce electrones provenientes de los seis ligandos de cloro. W está en el estado de oxidación +6 y es una especie d 0 que no aporta electones. ¿Puede la teoría de campos de ligandos explicar esta excepción de la regla de 18 electrones? Comencemos nuevamente con el diagrama MO considerando solo la unión σ-bonding. Los doce electrones ligando entran en los orbitales de unión 1a 1g, 1t u y 1e g. Los orbitales t 2g no enlazantes y 2e g antiadherentes permanecen vacíos debido a la ausencia de electrones d metálicos. Podemos ver que todos los orbitales moleculares enlazantes están llenos y todos los demás están vacíos, explicando la estabilidad de la molécula, y con ello la excepción de la regla de los 18 electrones. Ahora consideremos la unión π además. Un ligando cloro es un donador p típico que utiliza sus electrones 3p que están orientados adecuadamente para la unión p. Por lo tanto, aquí solo consideramos los orbitales del grupo ligando T 1g para la unión. Estos orbitales están llenos de electrones porque un anión cloruro tiene una subcapa 3p completa. La interacción de los T 2g LGO con los orbitales metálicos t 2g crea una unión t 2g MO y una anti-unión t 2g MO. Podemos ver que los electrones π del ligando ahora tienen una energía menor que sin las interacciones π-del metal. Por lo tanto, la unión p ha estabilizado aún más el complejo. En cierto modo, ahora podemos incluso decir que tenemos un complejo de 18 electrones porque cuando agregamos th 6 electrones π a los 12 electrones σ obtenemos 18 electrones de unión en general. Estos 6 electrones adicionales no se contabilizan en el conteo de electrones porque el conteo de electrones trata el enlace W-Cl como un enlace sencillo y solo considera las interacciones σ-entre W y Cl.
Complejo tetraédrico de un 4to periodo de Metal de Transición
A continuación, consideremos un complejo tetraédrico y veamos si la teoría del campo de ligandos puede explicarlo bien. El grupo de puntos para un tetraedro es T d, y necesitaremos la tabla de caracteres de este grupo de puntos (Fig. 7.1.32)

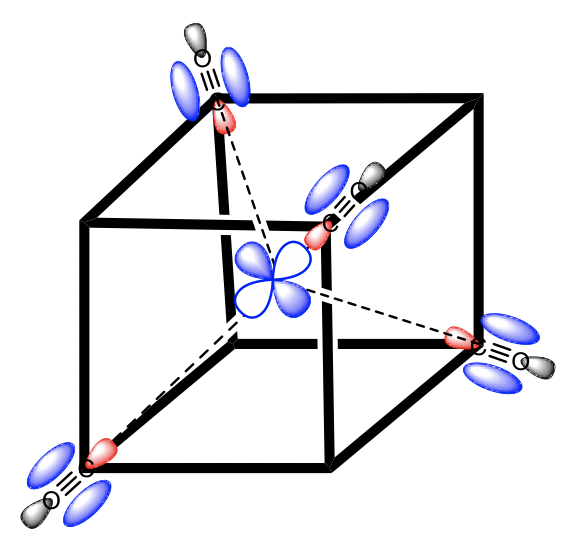
Elegimos el sistema de coordenadas como en la teoría del campo de cristal. Inscribimos el tetraedro en un cubo ocupando cada dos vértices del cubo con un ligando (Fig. 7.1.34). Entonces, dejamos que los ejes corran perpendiculares a las caras del cubo. A continuación, debemos pensar en los tipos de simetría de los orbitales de frontera metálica. Para un cuarto periodo los metales de transición son los orbitales 3d, 4s y 4p. Al mirar en la tabla de caracteres del grupo de puntos T d nos da sus tipos de simetría (Fig. 7.1.32 y Fig. 7.1.33). El orbital 4s tiene la simetría totalmente simétrica tipo A 1, encontramos las letras x, y, y z entre paréntesis en la representación irreducible del tipo T 2, y esto significa que los orbitales 4p están triplicamente degenerados y tienen la simetría tipo T2. Los orbitales 3d xy, 3d yz y 3d xz se encuentran en la misma representación irreducible, también son triples degenerados y tienen simetría T 2. Los orbitales 3d x 2 -y 2 y 3d z 2 tienen el tipo de simetría E según la tabla de caracteres para T d.

Ahora tenemos que pensar en los ligandos. Debido a que debemos considerar primero la unión σ-bonding, tenemos que encontrar el HOMO del ligando adecuado para la unión σ-bonding. Podemos elegir de nuevo un ligando carbonilo como ligando de ejemplo, y en este caso los homOS de los ligandos CO se usarían para la unión a. Esto en realidad no es inmediatamente obvio. Si elegimos el sistema de coordenadas del metal y los ligandos para que fueran los mismos, entonces el eje del enlace CO, que anteriormente definimos como el eje z, no apuntaría hacia el metal, y no sería posible solapar σ-con un orbital metálico. Por lo tanto, debemos darle a cada ligando un sistema de coordenadas diferente con los ejes z apuntando hacia el metal (Fig. 7.1.34). Solo entonces, la molécula de CO apuntaría hacia el metal, y estaría orientada para hacer un enlace σcon el metal. Generalmente, al construir MO, los ligandos siempre deben estar orientados a que se maximice la unión. Este suele ser el caso cuando se maximiza la superposición orbital para la unión σ-bonding.
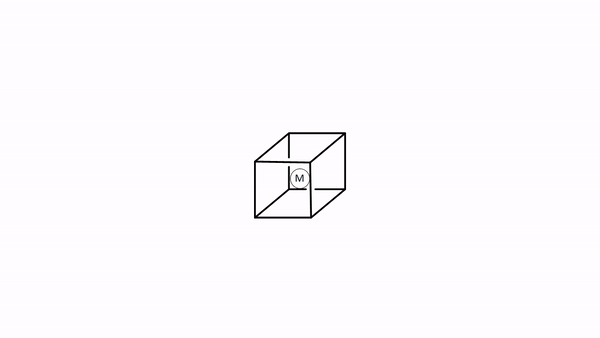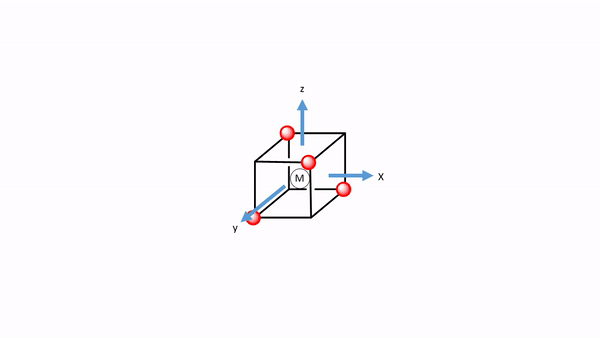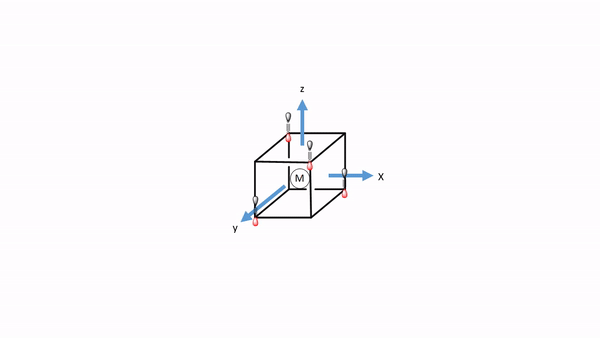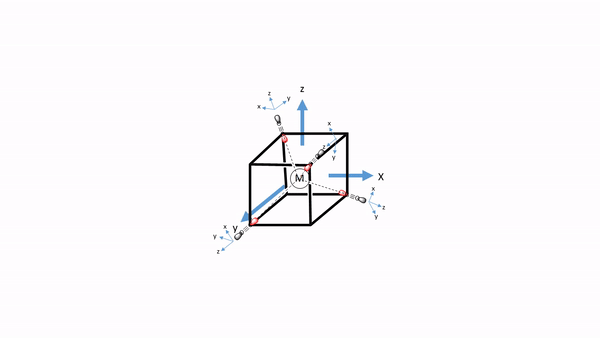
Figura 7.1.34 Un complejo tetraédrico inscrito en un cubo con ligandos de CO cada uno teniendo su propio sistema de coordenadas.
Debido a que tenemos cuatro ligandos tendremos cuatro homOS de ligandos que agruparíamos para formar cuatro orbitales de grupos de ligandos. ¿Qué tipos de simetría tienen? Para obtener los tipos de simetría necesitaríamos determinar primero la representación reducible, y luego las representaciones irreducibles por las que está compuesta esta representación reducible. No vamos a hacer esto explícitamente aquí y no pasaremos por todo el proceso matemático, y solo consideraremos los resultados.
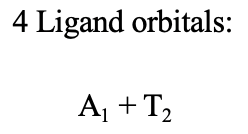
El resultado es que un grupo ligando orbital tiene la simetría tipo A1 y los otros tres tienen la simetría tipo T 2 (Fig. 7.1.35).
Ahora podemos construir el diagrama orbital molecular para el enlace σ-bonding (Fig. 7.1.36). Primero, podemos trazar los orbitales de frontera metálica de acuerdo con la energía del lado izquierdo del diagrama, y etiquetar los orbitales según sus tipos de simetría. En el lado derecho trazamos los cuatro orbitales del grupo de ligandos, y los etiquetamos según su tipo de simetría. Su energía debe ser aproximadamente la de los d-orbitales metálicos. Ahora podemos construir orbitales moleculares combinando orbitales atómicos y de grupos de ligandos del mismo tipo de simetría. El orbital 4s y un LGO tienen simetría A1, y por lo tanto podemos combinarlos para formar un orbital de unión 1a 1 y un orbital antiunión 2a 1. A continuación, dirijamos nuestra atención al tipo de simetría t 2. Los orbitales 4p y tres d metálicos tienen ese tipo de simetría, y así tienen tres LGO. Eso es en general nueve orbitales, por lo que debemos obtener nueve MO con simetría t 2. Debido a la triple degeneración, debe haber tres conjuntos de MO de triple degeneración. Podemos estimar que un conjunto se unirá, uno será aproximadamente no adherente y otro será anti-adhesión. Podemos etiquetarlos 1t 2, 2t 2 y 3t 2 respectivamente. El 2t 2 es en realidad algo antiadherencia en la naturaleza. Ahora solo quedan los e-orbitales metálicos. No encuentran pareja y permanecen sin vinculación.
Todavía necesitamos llenar los electrones en los MO. Los cuatro homOS de ligando se consideran completos, lo que da 4×2=8 electrones. Los electrones ocupan los orbitales de unión 1t 2 y 1a 1 explicando los cuatro enlaces dativos metal-ligando. Todos los electrones d metálicos, que podrían ser de hasta diez, necesitarían entrar en los orbitales e y/o t 2. Los orbitales e son los orbitales d z 2 y d x 2 -y 2 sin unión, y los orbitales 2t 2 son solo débilmente antienlaces y tienen un fuerte carácter orbital d-metal porque se han construido a partir del d xy , los orbitales d yz y d xz, y son bastante similares en energía a esos orbitales. Por lo tanto, podemos decir que los orbitales e y 2 t2 son los orbitales metálicos t 2 y e g en un campo de ligando tetraédrico. La diferencia de energía entre los orbitales e y 2t 2 es la energía de campo del ligando tetraédrico Δ T. Aquí se ve de nuevo la relación entre ligando y teoría de campos cristalinos. También se puede ver que la teoría del campo de ligandos puede explicar por qué la teoría del campo cristalino funciona como una teoría de unión aunque no sea una teoría de unión en realidad.
enlace π-en un complejo tetraédrico
¿Qué es el enlace π en un complejo tetraédrico?
Primero, tenemos que decidir si hay orbitales de ligando que estén adecuadamente orientados para superponerse con orbitales metálicos en forma π. Podemos ver que ningún ligando orbital se superpone con un metal d-orbital exactamente en forma π, sin embargo, los orbitales e π y 2e π*-de los ligandos aún se superponen de modo que en la unión se crea la interacción, y este orbital en general ocurre de manera similar a π-solapamiento. Por lo tanto, todavía podemos aproximar las interacciones de unión como π-bonding. Sin embargo, debemos considerar que debido al menor solapamiento orbital, el enlace π en los complejos tetraédricos es más débil que en los complejos octaédricos. ¿Cuántos orbitales de ligandos tendremos que considerar? Suponiendo que nos quedaremos con CO como ligando, habrá cuatro por ligando, y así habrá 4×4=16 en general. De estos, habrá ocho orbitales e π-orbitales de unión y ocho orbitales e π*-antiunión. Agrupamos los enlaces para formar un conjunto de ocho orbitales de grupos de ligandos y los antienlaces para formar otro conjunto de ocho orbitales de grupos de ligandos antiunión. ¿Cuáles serán sus tipos de simetría? Podemos determinar las representaciones reducibles e irreducibles para averiguarlo. Aquí no estamos pasando por el proceso exacto, sino que solo miramos los resultados. El resultado es que cada conjunto de orbitales de grupos de ligandos tiene dos orbitales de tipo E, tres orbitales de tipo T 1 y tres orbitales de grupo de ligandos de tipo T 2.
4_with_pi-bonding.gif)
Figura 7.1.38 Diagrama MO de un complejo tetraédrico de un metal de transición del 4º período (unión pi con aceptor pi)
Dado que ahora conocemos la simetría de los orbitales del grupo ligando, podemos combinarlos con orbitales d-metálicos de la misma simetría en el campo de ligandos tetraédricos para formar orbitales π-moleculares. Hagamos esto por el ejemplo del tetracarbonilo de níquel, un complejo tetraédrico de Ni en estado de oxidación 0 y cuatro ligandos carbonilo. Podemos usar el diagrama MO para la unión σ-que construimos previamente como punto de partida, y modificarlo para que tenga en cuenta la unión π (Fig. 7.1.38).
Primero, necesitamos agregar los nuevos orbitales del grupo de ligandos al diagrama MO. Solo necesitamos considerar los LGO E y T 2 y no los TGO T 1 porque ninguna órbita metálica tiene simetría t 1. A continuación, debemos tener en cuenta que el ligando CO es un ligando aceptor p fuerte. Esto quiere decir que solo necesitamos considerar los LGO formados a partir de los orbitales π*-antienlace. Debemos dibujar estos orbitales por encima de los LGO para la unión σ en el diagrama MO porque los LGO para la unión σse han formado a partir de los OMOs de CO mientras que los LGO antiunión para la unión π se han formado a partir de los LUMO de CO. Ahora podemos combinar los orbitales π-LGOS y d-orbitales metálicos de la misma simetría para formar π-MOS. Vemos que podemos combinar los LGO de tipo e con los orbitales de metal d z 2 y d x 2 -y 2 -y 2 de metal tipo e sin unión para formar un par de MO de unión de e-simetría, y un par antiunión de e-simetría, que podemos etiquetar 1e y 2e, respectivamente. Ahora, ¿cuál es el efecto en los π-LGOS tipo T 2? Primero tenemos que darnos cuenta de que ya tenemos tres conjuntos de t-MOS triple-degenerados generados a través de σ-interacciones. La interacción de los T 2 -LGO con los σ-t-2-MOS debe crear otro conjunto de orbitales triplemente degenerados para que el número total de orbitales con simetría t 2 siga siendo el mismo. La interacción ocurrirá principalmente entre los 2t 2 MO y los T 2 -LGO debido a que los 2t 2 MO tienen la energía más similar a los T 2 -LGO. Esto lleva a la disminución de la energía de los 2t 2 MOs, de manera que estos orbitales antiguamente débilmente antiadherentes se vuelven débilmente ligados. Los orbitales t 2 creados adicionalmente son débilmente antienlaces. Podemos etiquetarlos 3t 2. El anterior antienlace 3t 2 —orbital se volverá a etiquetar 4t 2. Podemos verificar dos veces que hemos construido los t 2 -MOS correctamente verificando que el número de T 2 -LGO, y eso incluye σ y π, más el número de orbitales T 2 metálicos es igual al número de orbitales moleculares t 2. Vemos que el número de orbitales T 2 metálicos + el número de T 2 LGO es 6+6=12. El número de t 2 MoS es 4×3 que también es 12.
Por último, todavía necesitamos llenar los electrones. Como se mencionó anteriormente, los π-LGOS están vacíos y por lo tanto el ligando no aporta ningún electrón al enlace π. El Ni se encuentra en el estado de oxidación 0 y así aporta 10 electrones porque Ni está en el décimo grupo de la tabla periódica. Debido a que un átomo de Ni neutro tiene la configuración electrónica 3d 8 4s 2 podemos dibujar ocho electrones en los orbitales 3d y dos electrones en los orbitales 4s. Los MOs 1t 1 y 1a 1 ya están llenos de electrones ligando debido a la unión σ-bonding. Así, los electrones metálicos deben entrar en el 2e y el 2t 2 MO. Tanto los orbitales e como los orbitales 2t 2 se están uniendo, y así podemos decir que los electrones d metálicos se han estabilizado debido a la naturaleza aceptora π del ligando. Vemos aquí una analogía al campo de ligandos octaédricos. Al igual que en el campo de ligandos octaédricos, los ligandos aceptores π tienden a disminuir la energía de los electrones d metálicos. También podemos preguntar: ¿Cuál es el efecto de un ligando aceptor π sobre Δ T? La respuesta es: Al igual que Δ o se hace más grande en el campo de ligando octaédrico, Δ T se hace más grande en un campo de ligando tetraédrico. Sin embargo, el incremento es mucho menor en comparación con el campo octaédrico. ¿Por qué? esto se debe a que en el campo de ligandos octaédricos, la energía de los orbitales e g no se ve afectada a través de las interacciones π-aceptor, y solo los orbitales t 2g se reducen en energía. En el caso del campo ligando tetraédrico se reduce tanto la energía de los orbitales e como del t 2, y la energía de los orbitales e se reduce solo ligeramente más que la energía de los orbitales t 2. Por lo tanto, la Δ T solo aumenta ligeramente. Este hecho también puede servir como una explicación adicional por qué los complejos tetraédricos nunca hacen complejos de bajo espín, incluso con ligandos aceptores de π fuertes. El efecto del ligando sobre Δ T es simplemente todavía demasiado pequeño debido a que tanto la energía de los orbitales e como del t 2 se han reducido.
Finalmente, discutamos los resultados en el contexto de la regla de los 18 electrones. Podemos ver que el número de electrones involucrados en la unión σ-se encuentra en la unión de orbitales moleculares. Así, se justifica decir que la teoría del campo ligando es capaz de explicar la regla de los 18 electrones.
Figura 7.1.39 Diagrama MO de un complejo tetraédrico de un metal de transición del 4º período (\(\pi\)-unión con\(\pi\) -donante)
Ahora pensemos en cómo un ligando donador de π influye en la magnitud de Δ T. En este caso solo necesitamos considerar los orbitales del ligando E y T2 de unión si el ligando tiene enlaces π. Esos orbitales estarán energéticamente por debajo de los LGO para el enlace σ-enlace. Si el ligando es un ion simple como el cloruro, entonces consideraríamos las OGs de los orbitales p rellenos que tienen simetría adecuada para el solapamiento π-. Esas LGO tendrían esencialmente la misma energía que las σ-LGO. Construyamos un diagrama MO cualitativo de TiCl 4 que tiene ligandos de cloro que son ligandos donadores típicos de π. Al construir el diagrama MO para la unión π-podemos comenzar nuevamente con el diagrama MO para la unión σ-y luego modificar este diagrama. Primero podemos graficar los π-T 2 y E g LGO rellenos con energía similar a la σ-LGO en el lado derecho del diagrama. Los orbitales del grupo de ligandos E ahora interactuarán con los orbitales de e-metal. Esa interacción conduce a un par de MO de unión de e-simetría, y un par de MO anti-unión de e-simetría. Esto significa que efectivamente, los orbitales de e-metal que antes no se unen se convierten en MO antiadhesión y se mueven hacia arriba en energía. El MO de unión tipo E se crea además y debe tener menor energía que el tipo E π-LGOS. La interacción de los T-2 -LGO con los orbitales t 2-metálicos crea un conjunto adicional de orbitales t2-orbitales de unión. Los orbitales 2t 2 se mueven hacia arriba en energía, y se vuelven más antiadherentes, como consecuencia de ello. Sólo podemos renumerar la t 2 y la e moS. Ahora todavía necesitamos llenar los electrones en los MO. Los π-LGOS tienen en general ocho electrones. Estos electrones entrarán en los orbitales moleculares de enlace 1e y 1t 2 recién formados. Vemos que esto lleva a una estabilización de estos electrones. Esto es una analogía con el campo ligando octaédrico. Cuando tenemos un ligando donador π, entonces ese el ligando π-electrones. ¿Qué pasa con los electrones metálicos? En TiCl 4 el Ti está en el estado de oxidación +4, por lo tanto es formalmente d 0, y no tiene electrones. Esto explica la estabilidad del TiCl 4 como complejo que no obedece a la regla de los 18 electrones. Tiene sólo 8 electrones, pero la situación de unión es sin embargo ideal. Todos los MO de unión están llenos, y todos los demás están vacíos. Si el Ti tuviera d electrones necesitarían entrar en los orbitales 2e y 3t 2 que son ambos antiadherentes. Esto desestabilizaría el complejo. En general, los ligandos donadores de π aumentan la energía de los electrones d metálicos, y esta es otra analogía con el campo de ligandos octaédricos. Ahora a la pregunta, ¿cuál es el efecto de un ligando donador de π en Δ T? Tanto los orbitales de frontera de metal e como t 2 han aumentado en energía, pero los orbitales electrónicos más que los orbitales t 2. Eso significa que el Δ T ha disminuido en general. Esta es una analogía adicional al campo de ligandos octaédricos. Los ligandos donadores de π conducen a una Δ T más pequeña.
En general, la teoría del campo de ligandos puede proporcionar nuevamente una explicación para la serie espectroquímica. Los donadores π-conducen a ligandos δ T más pequeños, y por lo tanto el complejo absorbe luz de longitudes de onda más grandes, los aceptores π conducen a una Δ T más grande y esos complejos absorben luz de longitudes de onda más pequeñas.
Complejo plano cuadrado de un metal de transición del 4to período
Como último ejemplo discutiremos ahora el diagrama orbital molecular de un complejo plano cuadrado. Este será el diagrama MO más complicado que discutiremos. La mayor complejidad proviene de la menor simetría en un complejo plano cuadrado. Pertenece al grupo puntual D 4h mientras que los complejos tetraédricos y octaédricos pertenecen a los grupos de puntos de alta simetría T d y O h.
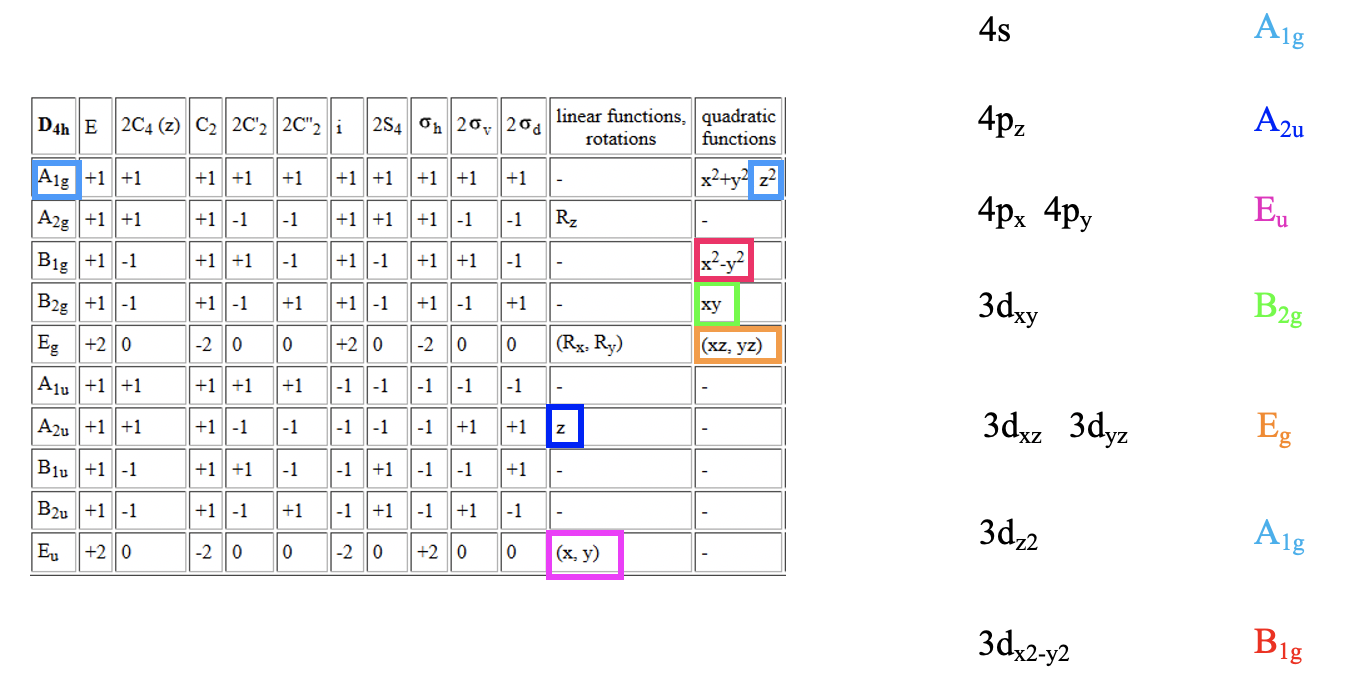
La menor simetría conduce a una reducción en la degeneración de los orbitales moleculares. Esto crea más niveles de energía y de ahí un diagrama orbital molecular más complicado. Pensemos a continuación en la definición de los ejes. Sería sensato definir el plano xy como el plano de la molécula con las coordenadas x e y pasando por los enlaces. El eje z se mantendría perpendicular al plano de la molécula. ¿Cuáles son los tipos de simetría de los orbitales de frontera metálica? Suponiendo un metal de transición del 4to periodo, los orbitales 3d, 4s y 4p serían los orbitales fronterizos. Al observar la tabla de caracteres del grupo de puntos D 4h (Fig. 7.1.10) se muestra que la simetría del orbital 4s es A 1g, la simetría de los orbitales 4p es A 2u para el orbital 4p z y e u para el 4p x y el 4p y orbitales. Los orbitales d tienen los tipos de simetría A 1g, B 1g, B 2g y E g para 3d z 2, d x 2 -y 2, 3d xy y 3d xz/3d yz, respectivamente.
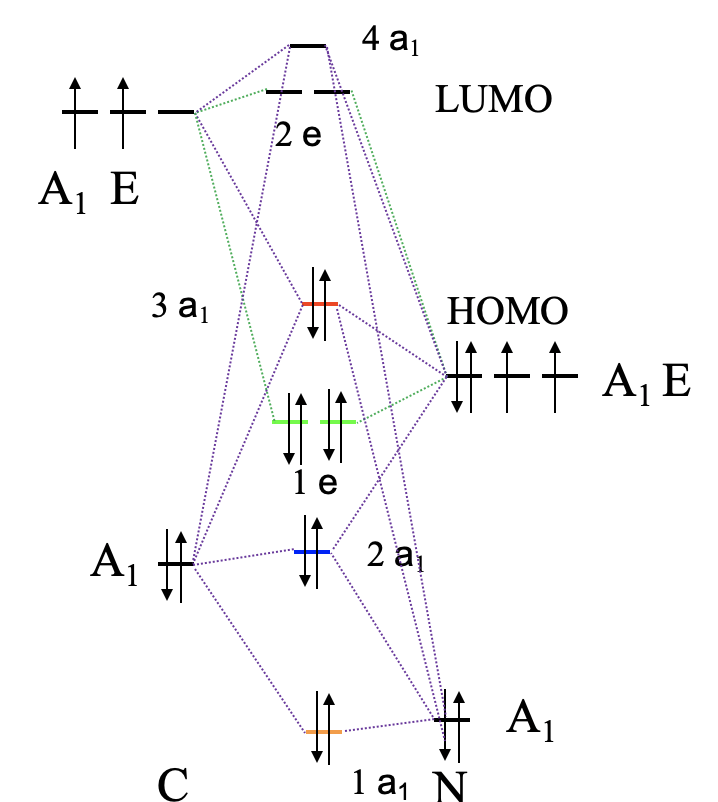
A continuación, se determina el ligando homOS adecuado para la unión σ. No hay complejos carbonílicos planos cuadrados, por lo tanto no elegiremos el ligando carbonilo como ligando de ejemplo aquí. En su lugar usaremos el ligando ciano relacionado. El ligando ciano es isoelectrónico con el ligando carbonilo. Formalmente, el O-átomo en CO es reemplazado por N. Debido a que N tiene un electrón menos que O, debemos agregar un electrón, dando al ligando ciano una carga 1-negativa. Al igual que en el CO, el ligando ciano tiene un triple enlace entre C y N, y hay un par solitario de electrones en C, y uno en N. El diagrama MO es similar al del CO, solo la diferencia de energía entre los orbitales C y N es algo menor que la de la diferencia de energía entre los orbitales C y O (Fig. 7.1.41). El número, la simetría y el orden energético de los MO de CN - y CO es el mismo. Por lo tanto, el CN - tiene los mismos HOOs adecuados para la unión σ-y podemos seleccionarlo para la construcción de σ-MOS. Cada ligando tiene un HOMO, por lo tanto tenemos en general cuatro HOMO.
Podemos agruparlos para formar LGO y determinar los tipos de simetría de los LGO. El resultado es que hay en el grupo ligando orbital con simetría A 1g, hay dos con simetría E u, y hay uno con simetría B 1g (Fig. 7.1.42).
Complejo Plano Cuadrado: σ-bonding
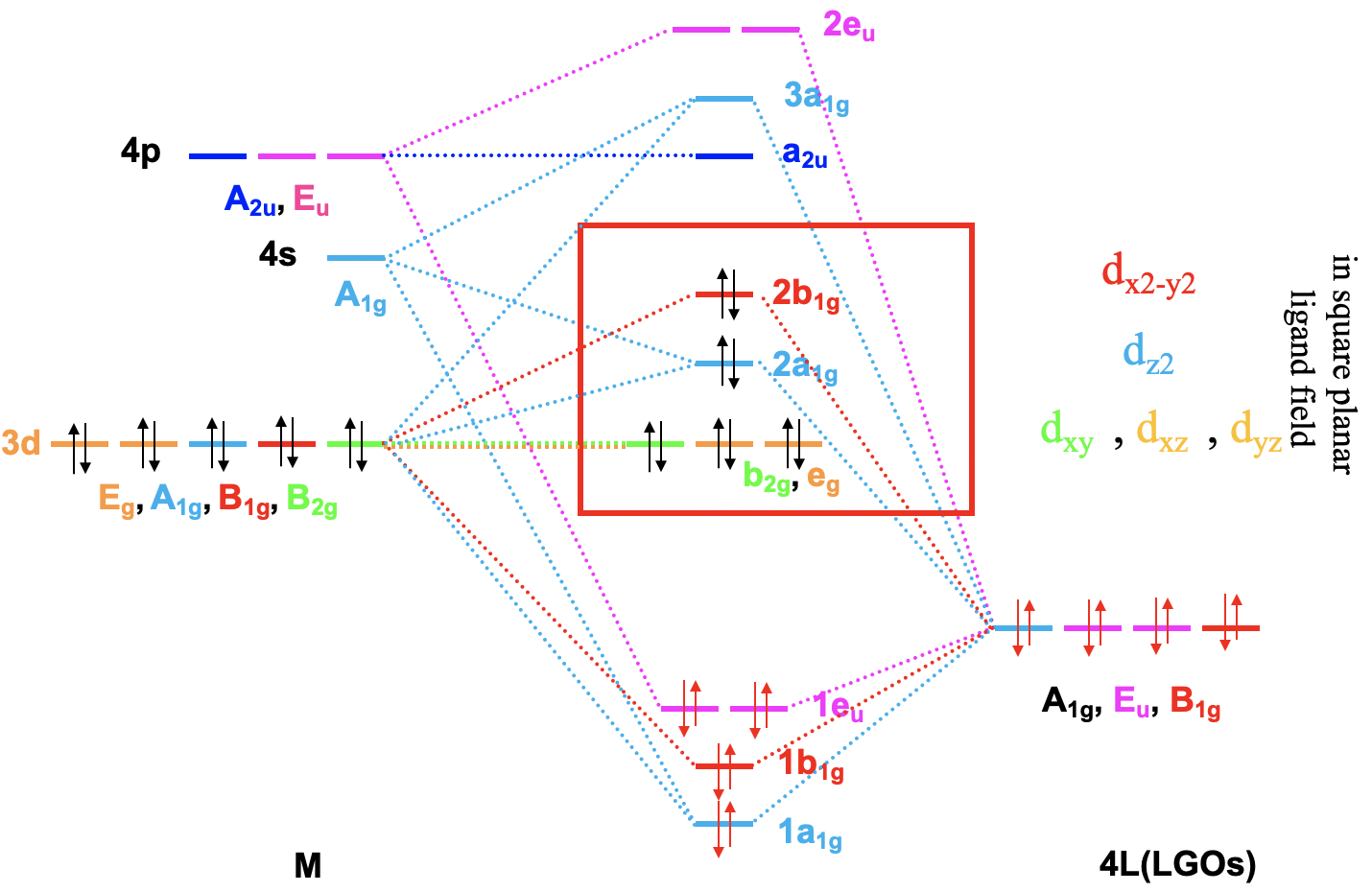
Ahora tenemos toda la información necesaria para construir un diagrama orbital molecular cualitativo para el enlace σ-bonding (Fig. 7.1.43). Como es habitual, primero trazamos los orbitales de frontera metálica a la izquierda y los orbitales del grupo de ligandos en el lado derecho del diagrama MO, y etiquetamos los tipos de simetría. Luego, necesitamos combinar orbitales de frontera metálica y grupos de ligandos del mismo tipo de simetría para formar orbitales moleculares. Podemos por ejemplo comenzar con la simetría tipo A 1g. Los orbitales 4s y los orbitales 3d z 2 tienen este tipo de simetría, y así lo tiene un grupo de ligandos orbitales. Por lo tanto, obtendremos tres orbitales moleculares de ese tipo de simetría, uno de enlace, uno aproximadamente no enlazante y un orbital molecular antiunión. Los trazamos de acuerdo con su energía esperada en el diagrama MO, y etiquetamos los MoS 1a 1g, 2a 1g y 3a 1g, respectivamente. A continuación, podemos observar los orbitales B 1g. El orbital 3d x 2 -y 2 y un LGO de ese tipo de simetría, y así podemos formar un MO de unión y un anti-adhesión a partir de esta combinación. Podemos ver además que podemos combinar los orbitales 4p x y 4p y que tienen simetría E u con los dos LGO restantes de la misma simetría. Esto da dos MO de unión doblemente degenerada de simetría e u, y dos uniones antiadherentes de la misma simetría. ¿Quedan orbitales con otros tipos de simetría? Sí, están los orbitales metálicos d xy, d xz, y d yz que tienen simetría B 2g, y E g, respectivamente, Además, existe el orbital p z que tiene una simetría 2u. Podemos ver que estos orbitales metálicos no encuentran pareja. No hay LGO de estos tipos de simetría. Por lo tanto, tenemos que dibujar estos orbitales como orbitales no vinculantes en el diagrama MO.
Ahora es el momento de llenar los electrones en los orbitales moleculares. Los LGO están ocupados con electrones porque se han formado a partir de los homOS del ligando. Debido a que tenemos cuatro LGO tenemos ocho electrones a considerar. Por lo tanto, estos electrones llenarán los orbitales moleculares 1a 1, 1b 1g y 1e u. Estos orbitales son todos orbitales moleculares enlazantes. Así, la teoría del campo de ligandos es capaz de explicar los cuatro enlaces y la forma plana cuadrada de la molécula. ¿A dónde irían los d-electrones metálicos? Podríamos tener hasta diez electrones d dependiendo del ion metálico, y estos diez electrones entrarían en los cinco MO de la siguiente energía superior. Estos son los orbitales b 2g y e g sin unión, el orbital 2a 1g aproximadamente no enlazante y el orbital 2b 1g antiunión. Podemos considerar estos orbitales como los orbitales metal-d en un campo de ligando plano cuadrado. Los orbitales b 2g es el orbital d z 2 en el campo de ligando plano cuadrado, los orbitales e g son d xz y d yz en un campo de ligando plano cuadrado, el orbital 2a 1g es el orbital d z 2 en a campo de ligando plano cuadrado y el orbital 2b 1g es el orbital d x 2 -y 2 en un campo de ligando plano cuadrado. Podemos ver que estos resultados son similares pero no completamente análogos a los que obtuvimos en la teoría del campo cristalino. Recuerde, que en la teoría del campo cristalino el orbital d x 2 -y 2 también había tenido la mayor energía. Sin embargo, el d z 2 fue menor en energía que el d xy, y el d xy no fue degenerado con los orbitales d xz y d yz. La diferencia se debe a que en la teoría del campo de ligandos el orbital d xy se considera no enlazante, no interactuando con los ligandos en absoluto, mientras que en la teoría del campo cristalino se asume una fuerte repulsión electrostática entre el d xy y los ligandos debido a que el d xy orbital está en el plano de la molécula.
Unión p Complejo Plano Cuadrado
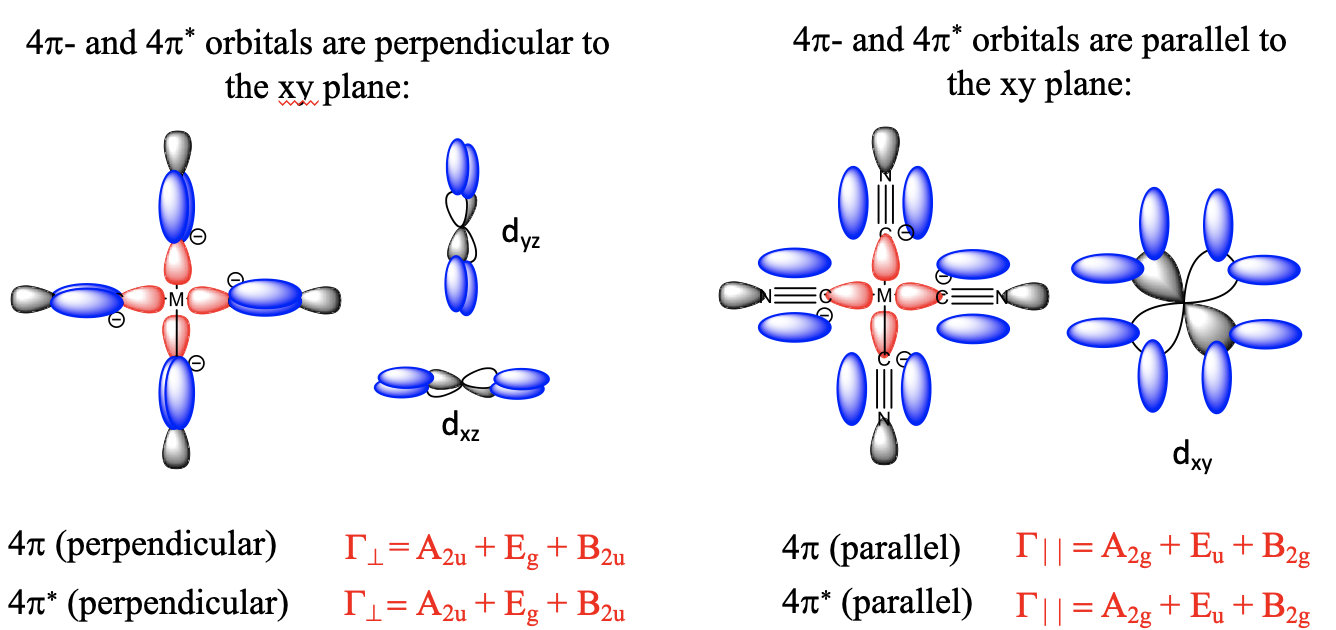
Ahora veamos la unión π de un complejo de metal de transición plano cuadrado (Fig. 7.1.44). Si asumimos un ligando CN -, entonces los orbitales que tienen simetría adecuada para superponerse con los orbitales de frontera metálica en forma π-son los orbitales 1e y 2e π y π* del diagrama MO de CO. Hay dos orbitales π y dos π* por ligando. Debido a que tenemos cuatro ligandos hay en general ocho orbitales π y ocho π*. Ahora debemos considerar que cuatro de los orbitales π-están en el plano xy, mientras que los otros cuatro están por encima y por debajo del plano xy. Llamamos a los primeros orbitales paralelos, y a los segundos orbitales perpendiculares, porque están orientados paralelos y perpendiculares al plano xy, respectivamente. Debemos distinguirlos porque tienen una simetría diferente y se superponen de manera diferente con los orbitales d metálicos. Esto se ilustra para los orbitales π de unión en la Fig. 7.1.44. Los antiligantes se comportan de manera similar. Por lo tanto, agrupamos los orbitales para formar cuatro conjuntos de orbitales del grupo ligando π, π ||, π* y π* ||. Nuestra siguiente tarea es determinar los tipos de simetría de estos orbitales. Nuevamente omitimos el proceso exacto, pero solo miramos los resultados. Tanto los orbitales π como los orbitales π* tienen un grupo de ligandos A 2u, dos E g y uno B 2u orbitales del grupo ligando. Los orbitales π || y π* || tienen un grupo de ligandos A 2g, dos E u y uno B 2g orbitales del grupo ligando.
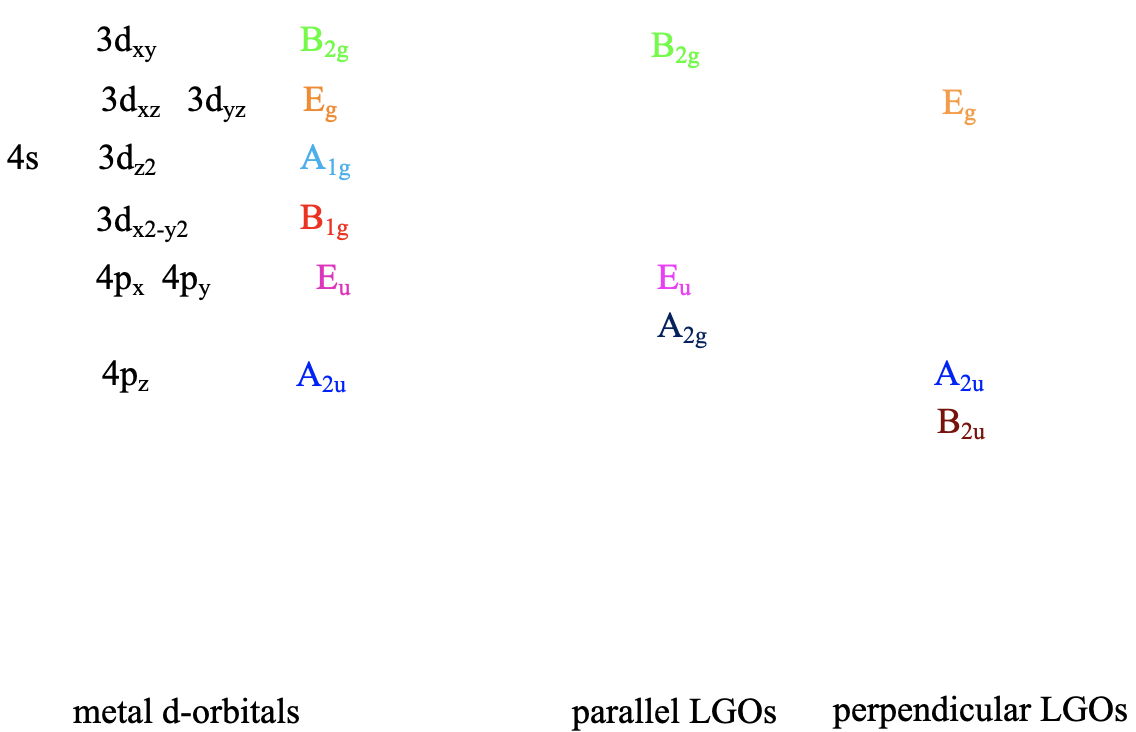
Ahora necesitamos agregar los orbitales π-moleculares al diagrama orbital molecular que habíamos construido anteriormente para dar cuenta de la unión σ-enlace. Primero, debemos considerar qué π-LGOS tienen la simetría correcta para producir π-MOS con orbitales d metálicos (Fig. 7.1.45). Podemos ver que entre las OGs paralelas los orbitales A 1g y B 1g encuentran socios, pero no los orbitales E u, Para los LGO perpendiculares los orbitales E g encuentran socios, pero no los orbitales A 2u y B 2u. Por lo tanto, solo necesitamos considerar las A 1g, las B 1g y las E g LGO. Para complicar las cosas, para obtener un diagrama MO realista para un complejo plano cuadrado con ligandos ciano es necesario considerar también las interacciones de los orbitales 4s y 4p con los π-LGOS. La simetría de los 4s fue A 1g y la simetría de los orbitales 4p fue A 2u y E u, respectivamente. El 4s todavía no encuentra pareja, pero los orbitales 4p sí.
Diagrama MO de un complejo plano cuadrado con un ligando ciano
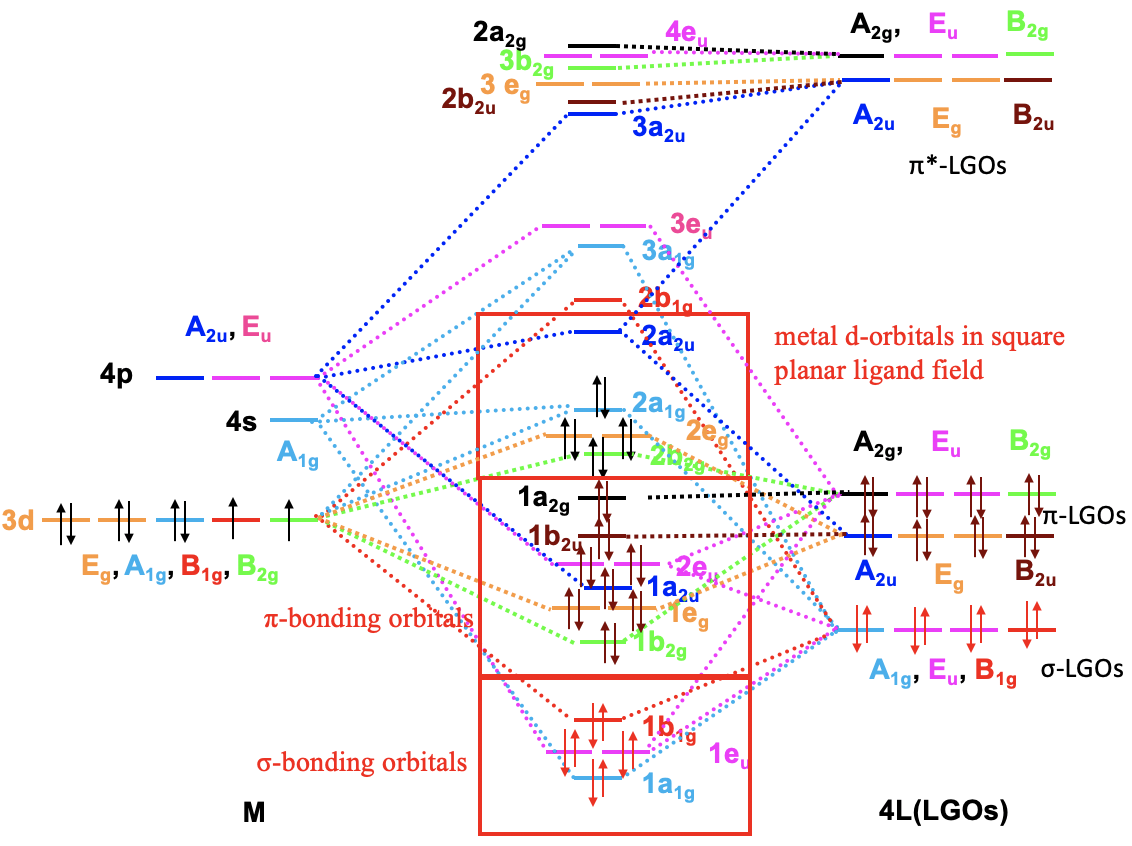
Construyamos ahora el diagrama MO de un complejo plano cuadrado con un ligando ciano considerando tanto el enlace σ como el enlace π para el ejemplo del anión complejo Ni (CN) 4 2-. Debido a la complejidad del diagrama no modificaremos el diagrama MO para la unión σ-bonding, como hicimos anteriormente, sino construiremos el diagrama MO completo desde cero (Fig. 7.1.46). Como antes podemos trazar los orbitales 3d, 4s y 4p metálicos en el lado izquierdo del diagrama. En el lado derecho del diagrama están ahora los LGO para σ así como π-bonding. Para el complejo Ni (CN) 4 2- los σ-LGOS están energéticamente algo por debajo de los d-orbitales metálicos, los π-LGOS de unión tienen aproximadamente la misma energía que los orbitales d-metálicos, y los π*-LGOS antiunión tienen una energía significativamente mayor que los orbitales 4p. Podemos construir nuevamente el tipo de simetría MoS por tipo de simetría hasta que se complete el diagrama MO. Podemos comenzar con la simetría tipo A 1g. Vemos que solo hay una σ-LGO que interactúa con los 4s y el 3d z 2 que también tienen simetría A 1g. Eso lleva a una unión, una aproximadamente no unión, y anti-unión un 1g MO que podemos etiquetar 1a 1g, 2a 1g, y 3a 1g, respectivamente. A continuación, podemos por ejemplo mirar los orbitales B 2g. Hay un d-orbital que tiene esa simetría, así como dos LGO. La interacción es casi solo con el enlace B 2g LGO porque las energías son similares. Esto conduce a una unión y una anti-unión π-MO con simetría B 2g. El LGO antiadherencia es demasiado alto en energía y no interactúa significativamente. Por lo tanto podemos dibujarla con la misma energía en el diagrama MO. Ahora vayamos a los orbitales B 1g. Hay un metal d-orbital con simetría B 1g, y un σ-LGO con simetría B 1g. Esto produce un MO de unión y anti-adhesión con simetría B 1g. Ahora vayamos a los orbitales E g. Hay dos E g d-orbitales metálicos, dos E g π-LGOS y dos E g π*-LGOS. Nuevamente, los π*-LGOS están demasiado alejados en energía, por lo que la interacción de unión es principalmente entre los orbitales metálicos los π-LGOS. Esto conduce a un par de MO de unión y un par de MO antiadherentes. Los π*-LGOS se pueden dibujar con energía inalterada en el diagrama MO. Los orbitales 4p del metal tienen tipos de simetría, aún no hemos considerado: A 2u y E u. Empecemos por los orbitales E u. Hay dos σ-e u LGO, dos π-e u -LGO y dos π*-e u LGO. En general tenemos cuatro pares de orbitales E U, y por lo tanto esperaríamos cuatro pares de e-u -MO. Podemos hacer la aproximación de que habrá uno de unión, uno débilmente ligado, uno débilmente antiadherentes y uno fuertemente antiadherentes. Podemos etiquetarlos 1e u, 2e u, 3e u y 4e u, respectivamente. El 4e u tiene casi solo contribuciones de los LGO π*-e U antienlace debido a argumentos energéticos, por lo tanto solo está conectado a estos a través de una línea punteada, y no a los otros orbitales E u. Ahora a los orbitales A 2u: Hay un π y un π*-orbital de simetría A 2u que se pueden combinar con los orbitales 4p A 2u. Esto conduce a tres MO de A 2u en general, una unión, una aproximadamente sin unión y una anti-unión. Ahora hemos utilizado todos los orbitales d metálicos. Notamos que todavía hay algunas OGL que no hemos utilizado. Estos son los π y π* A 2g y los B 2u LGO. Permanecen sin unión se pueden dibujar en consecuencia en el diagrama MO.
Ahora necesitamos llenar los electrones en el diagrama MO. En primer lugar, están los electrones σa considerar. En general, hay ocho electrones provenientes de los cuatro σ-LGOS ocupados. Podemos llenar estos ocho electrones en los cuatro MO con la energía más baja. Estos son el 1a 1, el 1e u y el 1b 1. Todos estos son MO de unión que explican los cuatro enlaces σ-dentro del complejo. Podemos llamar a estos cuatro orbitales los orbitales de unión σ. A continuación tenemos que considerar los electrones en los π-LGOS. En general hay ocho de estos orbitales que contienen 16 electrones. Debemos llenar estos electrones en los ocho MO de las energías siguientes superiores. Estos son los orbitales 1b 2g, 1e g, 1a 2u, 2e u, 1b 2u, y 1a 2g. Vemos que estos orbitales están enlazados o no enlazados, y así en general los electrones π del ligando se estabilizan debido a las interacciones π-con el ligando. Podemos llamar a estos orbitales los orbitales moleculares de enlace π. Por último tenemos que mirar a los electrones d metálicos. Asumimos Ni como el metal con Ni en el estado de oxidación +2 y configuración de electrones d 8. Necesitamos llenar estos ocho electrones en los cuatro siguientes MO de mayor energía. Esto llena los orbitales 2b 2g, 2e g, y 2a 1g, los cuales son no enlazantes o antiadherentes. Esto significa que los electrones metálicos están desestabilizados en general. Este comportamiento es consistente con el ciano-ligando que actúa como un ligando donador de π. Entendemos de esto por qué los complejos planares cuadrados prefieren 16 sobre 18 electrones y desobedecen la regla de 18 electrones. Si tuviéramos dos electrones más entonces estos necesitarían entrar en los orbitales 2a 2u energéticamente mucho más altos. La brecha de energía entre los orbitales 2a 2u y 2a 1g es significativamente mayor que entre los orbitales 1a 1g, 2b 2g y 2e g. En general podemos llamar a los orbitales 2a u, 2a 1g, 2e g y 2b g los orbitales d-orbitales metálicos en un campo de ligando plano cuadrado. En un campo de ligando plano cuadrado tenemos tres Δs. Δ 3 es la diferencia de energía entre los orbitales 2b 2g y 2e g, Δ2 es la diferencia de energía entre los orbitales 2e g y 2a 1g, y Δ1 es la energía entre los 2a 1g y los 2a 2 u orbitales. Δ 1 es mucho más grande que los otros dos. También casi siempre es mayor que la energía de apareamiento de espín, por lo tanto, los complejos planos cuadrados son casi siempre complejos de bajo espín.
En general, vemos que un diagrama MO puede llegar a ser muy complejo incluso para una molécula relativamente simple de simetría relativamente alta. Cabe señalar que para diagramas MO tan complejos como este, ya no es posible determinar el orden exacto de los niveles de energía solo por inspección cualitativa, y sin cálculos exactos. Sin embargo, se puede ver que es posible entender fácilmente un diagrama MO complejo siguiendo el enfoque de combinación lineal adaptada a simetría de orbitales atómicos. Solo necesitamos pasar paso a paso por los tipos de simetría y construir MOs combinando orbitales del mismo tipo de simetría hasta que el proceso esté completamente. Ya hemos llegado al final del capítulo sobre teoría de campos de ligandos.