
4.1: Cinética y Mecanismos-
- Page ID
- 76915

Una de las fuentes más poderosas de evidencia de cómo procede una reacción particular proviene del estudio de las velocidades de reacción o la cinética de reacción. En tales estudios, cómo cambia la velocidad de una reacción [1] se mide en función de la concentración de cada reactivo. Una de las formas más comunes de medir este cambio es mediante el uso de una técnica espectroscópica. Por ejemplo, si el compuesto absorbe en la región UV-VIS del espectro, la absorbancia es proporcional a la concentración. Por lo tanto, si la concentración de la sustancia cambia, se puede medir por cambios en la absorbancia. La reacción se lleva a cabo varias veces con todos menos uno de los reactivos establecidos como constantes; luego se agrega una concentración diferente del reactivo restante y se mide la velocidad de la reacción. Esto se repite para cada reactivo en varias concentraciones. El resultado final de dicho estudio es producir lo que se conoce como la ecuación de velocidad; para una reacción genérica\(\mathrm{A}+\mathrm{B} \rightarrow \mathrm{C}+\mathrm{D}\) la ecuación de velocidad toma la forma:\[\text { Rate }=k[\mathrm{A}]^{x}[\mathrm{B}]^{y}\]
En esta ecuación,\(k\) está la constante de velocidad, y los exponentes x e y nos hablan de cómo la concentración de cada reactivo influye en la velocidad. La suma de los exponentes (es decir,\(x+y+\ldots\)) es el orden de la reacción. Por ejemplo, si\(x = 1\), entonces la tasa está directamente relacionada con la concentración de\(\mathrm{A}\). Si ambos\(x\) y\(y = 1\), entonces la velocidad es directamente proporcional a ambos\([\mathrm{A}]\) y\([\mathrm{B}]\), y el orden general de reacción es = 2. Si un exponente = 0 entonces la velocidad no depende de esa concentración de reactivo, y ese reactivo se puede eliminar de la ecuación de la ley de velocidad (ya que\([n]^{0}=1\) no importa cuál sea el valor de concentración de n).
La idea más importante a recordar es que la ecuación de velocidad solo contiene los reactivos que están involucrados en el paso determinante de la velocidad (es decir, el paso más lento) de la reacción. Si la reacción procede por una serie de pasos, entonces el paso con la mayor energía de activación será determinante de la velocidad, y solo aquellos reactivos que participen en este paso estarán presentes en la ley de velocidad. [2] Dado que la ley de tasas se determina empíricamente, la ley de tasas nos proporciona evidencia sobre el mecanismo de la reacción.
Evidencia para el\(\mathbf{S}_{\mathbf{N}} 2\) Mecanismo:
La reacción que discutimos anteriormente en el curso se conoce como una\(\mathrm{S}_{\mathrm{N}} 2\) reacción que es taquigrafía de S ubstitución, N nucleofílica, Segundo orden. Propusimos un mecanismo para esta reacción sin aportar ninguna evidencia empírica, pero ahora vamos a usar algo de lo que ha aprendido para considerar más cuidadosamente la evidencia para este mecanismo.
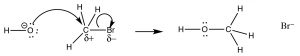
La reacción es de segundo orden:
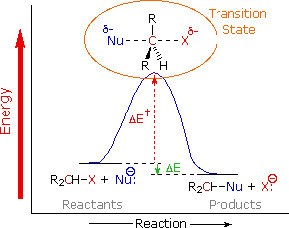
la primera prueba proviene de la ley de tasa cinética. La velocidad de reacción depende tanto de la concentración del sustrato como del nucleófilo:\(\text { rate }=k[\mathrm{RX}][\mathrm{Nu}]\). Esto significa que ambos deben estar presentes en el paso de determinación de la tasa. La explicación más simple que concuerda con este hallazgo es la que ya hemos propuesto: el nucleófilo ataca al carbono electrófilo al mismo tiempo que sale el grupo saliente. Es decir, la reacción tiene lugar en un paso continuo. Un diagrama de energía de reacción en el que trazamos Energía v progreso de reacción se ve así (\(\rightarrow\)). En esta reacción, solo hay una barrera energética, solo un máximo en la vía de reacción. La energía de esta barrera se conoce como la energía de activación\(\Delta \mathrm{E}_{+}^{+}\). Al observar el diagrama de reacción, también observamos que la reacción es exotérmica (o exergónica si trazamos energía de Gibbs), ya que la\(\Delta \mathrm{E}\) de la reacción general es negativa). La especie en el pico de la barrera de energía de activación se conoce como el estado de transición, y su estructura y energía asociada determina la velocidad de la reacción.
La estructura del sustrato afecta la tasa:
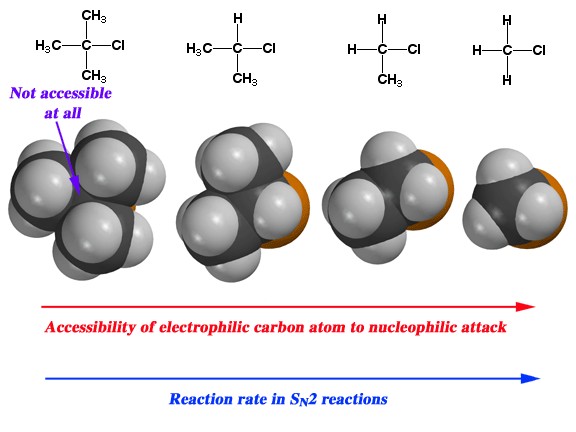
Quizás haya notado en nuestras discusiones anteriores sobre la sustitución nucleofílica que el sustrato orgánico siempre fue un metilo o carbono primario unido a un buen grupo saliente. La razón fue que en las reacciones que hemos considerado, tanto la velocidad como el mecanismo de reacción son altamente dependientes de la estructura del sustrato. A medida que aumenta el número de grupos metilo unidos al carbono primario (de 0 para un grupo metilo en sí a tres [terciarios]) la velocidad de reacción se ralentiza, como se muestra. La velocidad de\(\mathrm{S}_{\mathrm{N}} 2\) reacción para un sustrato terciario es insignificante.
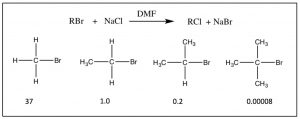
Entonces surgen dos preguntas: primero, ¿por qué son diferentes estas velocidades de reacción? y segundo, ¿por qué este cambio en las velocidades de reacción es evidencia del\(\mathrm{S}_{\mathrm{N}} 2\) mecanismo? Ambos pueden ser respondidos analizando más de cerca la reacción desde una perspectiva molecular. Recuerde, todos los reactivos se disuelven en un solvente; el movimiento térmico lleva a que colisionen entre sí y con moléculas de solvente. Para que el nucleófilo y el sustrato reaccionen entre sí, primero tienen que chocar entre sí. Para que ocurra una reacción, esa colisión tiene que transferir suficiente energía para que el complejo (sustrato + nucleófilo) pueda formar el estado de transición; además, para formar la molécula de estado de transición, las moléculas deben colisionar entre sí en la orientación correcta. Una vez formado, el estado de transición puede decaer para formar los productos de la reacción.
Recordemos que nuestra estructura propuesta para el estado de transición para esta reacción tiene el carbono central conectado a cinco grupos: el nucleófilo entrante, el grupo saliente y los otros tres sustituyentes que no cambian durante la reacción (no son parte de la reacción). A medida que se forma el enlace entre el nucleófilo y el carbono del sustrato, y el enlace se rompe entre el carbono y el grupo saliente, el carbono cambia su estado de hibridación. ¿Qué significa eso? En la molécula sustrato, el carbono de reacción se une a grupos circundantes (\(\mathrm{H-}\)o\(\left.\mathrm{CH}_{3}-\right)\)) con enlaces formados a partir de\(\mathrm{sp}^{3}\) orbitales. En el estado de transición, este carbono sigue unido a aquellos grupos que permanecerán en la molécula producto, pero ahora con enlaces formados a partir de\(\mathrm{sp}^{2}\) orbitales. Adicionalmente, todavía está unido tanto al grupo saliente como al nucleófilo entrante usando un orbital p para formar estos enlaces parciales. Se puede pensar en este proceso como la densidad de electrones que se canaliza desde el nucleófilo a través del carbono y por el otro lado hacia el grupo saliente. Sin embargo, para que esto ocurra el nucleófilo solo puede comenzar a enlazarse cuando se acerca desde la parte posterior del enlace al grupo saliente\(\rightarrow\).
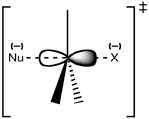
En este punto, bien podrías encontrarte preguntándote: ¿qué tiene que ver todo esto con la estructura del sustrato? Para que ocurra una reacción, las únicas colisiones productivas son aquellas en las que el nucleófilo comienza a formar un enlace con la parte posterior del orbital\(\mathrm{sp}^{3}\) híbrido; pero la estructura del sustrato influye en la probabilidad de tal evento. En los sustratos terciarios (por ejemplo\(\left(\mathrm{CH}_{3}\right)_{3} \mathrm{CBr}\)) la aproximación al sustrato se ve obstaculizada por los grupos alquilo voluminosos de tal manera que la probabilidad de que el nucleófilo interaccione con el centro reactivo es baja. Este fenómeno se denomina impedimento estérico y proporciona una explicación del orden de reacción de\(\mathrm{S}_{\mathrm{N}} 2\) las reacciones.
\(\mathrm{S}_{\mathrm{N}} 2\)reacciones en un centro quiral:
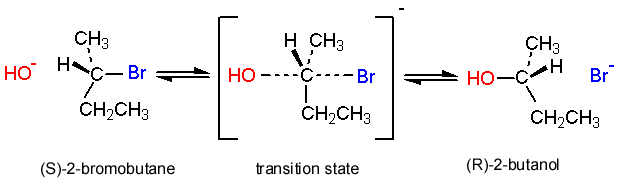
Otra pieza de evidencia del\(\mathrm{S}_{\mathrm{N}} 2\) mecanismo es lo que sucede cuando una\(\mathrm{S}_{\mathrm{N}} 2\) reacción tiene lugar en un centro quiral (dentro de una molécula). Resulta que la configuración en ese centro se cambia; el carbono se invierte (como un paraguas que sopla del revés en el viento) de manera que un enantiómero S se convierte en un enantiómero R. De hecho, es posible seguir el progreso de una reacción de S N 2 que involucra un centro quiral usando un polarímetro (el instrumento utilizado para medir la actividad óptica); a medida que la reacción avanza hasta su finalización, la rotación óptica de la solución cambia con el tiempo. Para cada sustrato en particular, la dirección y magnitud de la rotación para el producto serán diferentes. Este fenómeno se llama inversión Walden y proporciona otra pieza de evidencia para apoyar el mecanismo de reacción propuesto.
El papel del disolvente en una\(\mathrm{S}_{\mathrm{N}} 2\) reacción:
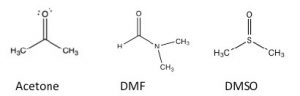
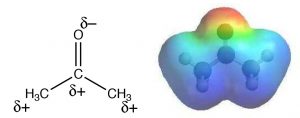
\(\mathrm{S}_{\mathrm{N}} 2\)las reacciones generalmente se llevan a cabo en un solvente (¿por qué es eso?). Estudios empíricos revelan que tales reacciones proceden más rápidamente cuando se llevan a cabo en lo que se conoce como disolvente aprótico polar. Entonces, ¿qué es un solvente aprótico polar? El término significa que el disolvente es polar pero sin protones ácidos. Ejemplos de disolventes apróticos polares son acetona, dimetilformamida (DMF) y dimetilsulfóxido (DMSO): cada uno es polar, pero carece de un protón potencialmente ácido como el H que está unido al\(\mathrm{O}\) en etanol\(\mathrm{CH}_{3} \mathrm{CH}_{2} \mathrm{OH}\) o en agua\(\mathrm{H-O-H}\). El agua (y metanol y etanol) es un disolvente prótico polar. En un disolvente aprótico polar, el extremo negativo del\(\mathrm{C=O}\) o\(\mathrm{S=O}\) dipolo se localiza en el\(\mathrm{O}\), mientras que el extremo positivo es difuso y deslocalizado. Por ejemplo, en acetona, el oxígeno tiene una\(\delta-\) carga sobre el oxígeno mientras que la carga positiva del dipolo se deslocaliza sobre ambos grupos\(\mathrm{C}\) y metilo como se muestra en el mapa de potencial electrostático (\(\rightarrow\)). En la práctica, los solventes apróticos polares pueden solvatar bien los cationes a través de interacciones con el extremo negativo localizado del dipolo, pero no pueden solvatar muy bien los aniones.
Recordemos que la solvatación es una interacción que disminuye la energía del sistema, haciéndolo más estable (menos reactivo). Por lo tanto, un disolvente que deje al nucleófilo (el anión) sin solvatar lo hará más reactivo. En contraste, un disolvente prótico polar (como el agua o el etanol) puede solvatar al nucleófilo, a través de interacciones con el extremo positivo de un dipolo que se localiza en un ácido\(\mathrm{H}\), estabilizando al nucleófilo y haciéndolo menos reactivo. En resumen,\(\mathrm{S}_{\mathrm{N}} 2\) las reacciones ocurren en un solo paso con inversión en un centro quiral. Tales reacciones son generalmente más rápidas para sustratos sin obstáculos y se aceleran cuando se llevan a cabo en disolventes apróticos polares.