
4.2: El\(\mathrm{S}_{\mathrm{N}} 1\) Reaction
- Page ID
- 76916

Si ya has tomado un curso de laboratorio de química, sin duda has observado que no obtienes un rendimiento del 100% para una reacción en particular y muchas veces se genera más de un producto. Este no es (generalmente) un caso de técnica experimental defectuosa, sino que refleja la complejidad de los sistemas de reacción. Dada nuestra experiencia en el uso de evidencia para apoyar los mecanismos de reacción propuestos, echemos un vistazo a otro conjunto de condiciones para las sustituciones nucleofílicas. Considera la reacción:
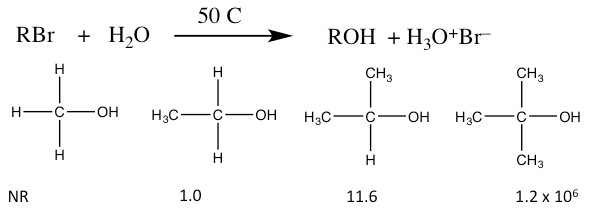
Tasas Relativas de\(\mathrm{S}_{\mathrm{N}} 1\) Reacciones
En este escenario, el agua es tanto el nucleófilo como el solvente. El agua no es un nucleófilo muy fuerte y es un solvente prótico. En estas condiciones, se produce una sustitución nucleofílica, pero esta reacción difiere en varias formas empíricamente observables de las\(\mathrm{S}_{\mathrm{N}} 2\) reacciones discutidas anteriormente.
- La velocidad de reacción depende únicamente del sustrato. La reactividad del nucleófilo es irrelevante. La ecuación de velocidad para estas reacciones es:\(\text { rate }=k[\mathrm{RBr}]\). La reacción es de primer orden, por lo que se denomina\(\mathrm{S}_{\mathrm{N}} 1\) reacción (Sustitución, Nucleofílica, Primer Orden).
- Las velocidades relativas de reacción para los sustratos se invierten de la\(\mathrm{S}_{\mathrm{N}} 2\) reacción. Es decir, la velocidad de reacción es mayor para la terciaria y la más baja para la forma primaria.
- Cuando un centro quiral experimenta una\(\mathrm{S}_{\mathrm{N}} 1\) reacción, el producto contiene una mezcla de ambos enantiómeros posibles, en lugar de la inversión de configuración encontrada con\(\mathrm{S}_{\mathrm{N}} 2\) las reacciones.
- La reacción es acelerada por solventes próticos polares (que ralentizan\(\mathrm{S}_{\mathrm{N}} 2\) las reacciones).
¿Cómo podemos explicar estos comportamientos? Nuestra suposición es que está involucrado un mecanismo de reacción diferente; en comparación con las\(\mathrm{S}_{\mathrm{N}} 2\) reacciones que hemos estado considerando. ¿Cuál podría ser ese mecanismo?
Dado que la velocidad de reacción observada depende únicamente de la concentración de sustrato (1) podemos suponer que solo la molécula sustrato está involucrada en la etapa de determinación de la velocidad. Entonces, ¿cómo comienza la reacción? Debemos asumir que la reacción implica romper el enlace (ya que no hay nada más que pueda suceder si solo hay una molécula en el paso determinante de la velocidad. Dado que la ruptura del enlace requiere energía, las colisiones térmicas con moléculas de disolvente deben impulsar este evento de ruptura de enlaces. Sin embargo, también sabemos que la molécula del solvente no participa en este paso de la reacción (porque no está en la ley de velocidad). Una posibilidad es que el enlace al grupo lábil se rompa, dando como resultado un carbono cargado positivamente (un carbocatión) y el anión del grupo lábil\(\rightarrow\). Esta es la primera instancia que hemos visto de un carbocation. La formación de este carbocatión requiere energía (ya que el enlace se está rompiendo), y podemos inferir que tiene una alta energía de activación. Los carbocationes en sí mismos son especies de alta energía que son muy reactivas. Sin embargo, son diferentes de los estados de transición en que es posible generar y detectar la carbocatión, generalmente tiene una vida corta, pero podemos detectar su presencia por métodos espectroscópicos. Esto es diferente de un estado de transición, que es la especie de mayor energía en el perfil de energía de reacción. Los estados de transición existen para una sola vibración molecular y tienen tiempos de vida en el orden femtosegundos.

Podríamos esperar que tal carbocatión reaccione rápidamente con cualquier nucleófilo potencial presente, que en este caso es la molécula de agua solvente.
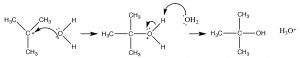
Aunque el agua es un nucleófilo pobre, reaccionará con el carbocatión altamente reactivo para dar la forma protonada intermedia. A esto le sigue una transferencia de protones a otra molécula de agua (disolvente) para formar el producto final.
Tal mecanismo satisface nuestras observaciones experimentales. Tiene un primer paso lento (limitación de velocidad, alta activación, que requiere energía) y un segundo paso más rápido (menor energía de activación). Debido a las diferencias en las energías de activación de los dos pasos, solo el primer paso está involucrado en la determinación de la velocidad de reacción general. Para averiguar cuál es el paso determinante de la tasa podemos ver que\(\Delta \mathrm{G}^{\dagger}\) para el paso 1 es mayor que\(\Delta \mathrm{G}^{\dagger}\) para el paso 2.
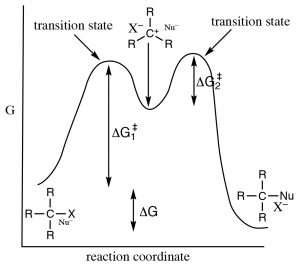
¿Por qué importa la estructura del sustrato?
La formación de carbocatión intermedio es la etapa determinante de la velocidad en dicha reacción, y por lo tanto se deduce que la estructura del carbocatión tiene un impacto en la energía de activación de la reacción, y por lo tanto su velocidad. Cuanto más estable es el carbocatión, menor es la energía del estado de transición que conduce al carbocatión. [3] Los factores que estabilizan el carbocatión también estabilizarán el estado de transición y disminuirán la energía de activación.
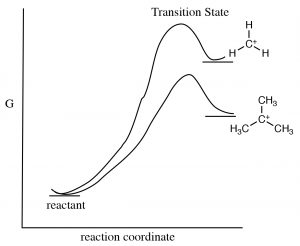
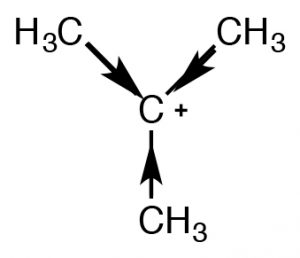
Existen dos mecanismos que pueden estabilizar los carbocationes y ambos predicen que, como el número de grupos alquilo unidos al\(\mathrm{C+}\) incremento, también lo hará su estabilidad. Un mecanismo que ya hemos encontrado es la inducción. Los grupos alquilo unidos al carbono central, son polarizables y su densidad electrónica es atraída hacia la carga positiva del carbono central deslocalizando así la carga positiva sobre los grupos alquilo. Cuantos más grupos alquilo unidos al carbono central, más pronunciada es esta estabilización. El segundo mecanismo, conocido como hiperconjugación, también deslocaliza la carga positiva. En la hiperconjugación, la densidad electrónica de cualquier\(\mathrm{C-C}\) enlace adyacente\(\mathrm{C-H}\) o enlace puede superponerse con el orbital p vacío en el carbocatión\(\mathrm{sp}^{2}\) hibridado, formando una especie de enlace pi y, nuevamente, deslocalizando la carga positiva sobre el resto de la molécula. Cuantos más grupos alquilo presentes (unidos al\(\mathrm{C+}\)), más pronunciado será este efecto.
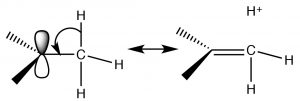
En conjunto, tanto la inducción como la hiperconjugación explican por qué una\(\mathrm{S}_{\mathrm{N}} 1\) reacción avanza más rápido con sustratos terciarios. El carbocatión terciario es más estable (relativo a los carbocationes secundarios y primarios) de manera que la reacción tiene una menor energía de activación.
¿Por qué los centros quirales racemizan?
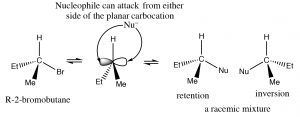
La respuesta a esta pregunta radica en la estructura del carbocatión. Es una estructura plana,\(\mathrm{sp}^{2}\) hibridada, simétrica. Una vez formado, puede ser atacado desde cualquier lado por un nucleófilo; en compuestos simples en qué lado se atacará el carbocatión implica un evento de colisión aleatoria, dando una mezcla de enantiómeros.
¿Por qué\(\mathrm{S}_{\mathrm{N}} 1\) las reacciones son aceleradas por solventes próticos polares?
Recuerde que un disolvente prótico polar (como agua o etanol) contiene un dipolo: un dominio parcialmente positivo y parcialmente negativo.
El ataque a un centro quiral da una mezcla racémica de productos.
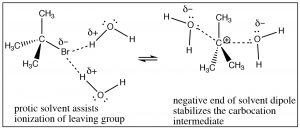
Este dipolo de molécula solvente cumple dos funciones: puede solvatar el grupo lábil, en efecto ayudando a eliminarlo del carbocatión a través de interacciones con el extremo positivo del solvente dipolo y puede solvatar el carbocatión a través de interacciones con el dominio negativo del dipolo solvente. En esencia, el disolvente ayuda en la ionización del grupo lábil, y disminuye la energía del carbocatión intermedio.
\(\mathrm{S}_{\mathrm{N}} 1\)reacciones en sistemas estabilizados por resonancia:
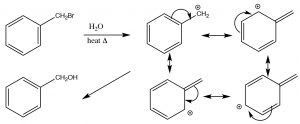
Como hemos visto,\(\mathrm{S}_{\mathrm{N}} 1\) las reacciones tienden a ocurrir cuando se puede formar un intermedio de carbocatión estabilizado. Además de aquellos cationes terciarios discutidos anteriormente, que se estabilizan por inducción e hiperconjugación, también hay ocasiones en las que incluso se pueden formar carbocationes primarios si hay una manera de estabilizarlos. Por ejemplo, cualquier carbono primario que pueda conjugarse con un sistema pi puede estabilizarse por resonancia. Por ejemplo, un carbocatión bencílico (es decir, cualquier carbono unido a un anillo de benceno) se puede estabilizar deslocalizando la carga positiva en el anillo de benceno. Esto disminuye la energía de activación para la reacción y hace posible una\(\mathrm{S}_{\mathrm{N}} 1\) reacción.
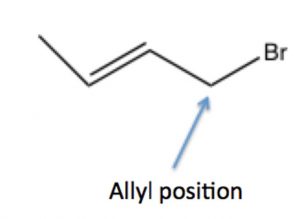
Un fenómeno similar puede ocurrir en sustratos con grupos salientes en posición alílica: es decir, en el carbono próximo a un doble enlace, donde el carbocatión resultante puede estabilizarse por resonancia por deslocalización en el sistema pi del doble enlace. Discutiremos este fenómeno con más detalle más adelante (en Capítulo\(7\)).
\(\mathrm{S}_{\mathrm{N}} 1\)o\(\mathrm{S}_{\mathrm{N}} 2\)?
Antes de seguir adelante, revisemos lo que sabemos\(\mathrm{S}_{\mathrm{N}} 1\) y\(\mathrm{S}_{\mathrm{N}} 2\) las reacciones. Si bien puede parecer un poco confuso, hay una serie de factores que pueden ayudarnos a predecir cuál podría ser el mecanismo potencial para una reacción, y también para predecir el producto. A medida que avanzamos hacia sistemas de reacción más complejos, será importante recordar que normalmente hay más de una vía que puede tomar una reacción, pero al comprender cómo ocurren las reacciones podemos ajustar las condiciones para que el producto que queremos sea el producto principal. En la siguiente tabla se resume lo que hemos comentado hasta ahora con respecto\(\mathrm{S}_{\mathrm{N}} 1\) y\(\mathrm{S}_{\mathrm{N}} 2\) reacciones.
| \(\mathrm{S}_{\mathrm{N}} 1\) | \(\mathrm{S}_{\mathrm{N}} 2\) | |
|---|---|---|
| Sustrato | \ (\ mathrm {S} _ {\ mathrm {N}} 1\)” style="height:14px; ">Terciario > secundario > bencílico~alílico
> primario > metilo |
\ (\ mathrm {S} _ {\ mathrm {N}} 2\)” style="height:14px; ">Metilo > primario > secundario> terciario |
| Grupo de salida | \ (\ mathrm {S} _ {\ mathrm {N}} 1\)” style="height:15px; ">Los buenos grupos de salida aumentan la tasa al disminuir la energía del TS | \ (\ mathrm {S} _ {\ mathrm {N}} 2\)” style="height:15px; ">Los buenos grupos de salida aumentan la tasa al disminuir la energía del TS Acelerado por nucleófilos fuertes |
| Nucleófilo | \ (\ mathrm {S} _ {\ mathrm {N}} 1\)” style="height:15px; ">Nucleófilos básicos no (Bronsted), nucleófilos débiles (bases fuertes promueven la eliminación—ver siguiente sección) | \ (\ mathrm {S} _ {\ mathrm {N}} 2\)” style="height:15px; "> |
| Solvente | \ (\ mathrm {S} _ {\ mathrm {N}} 1\)” style="height:15px; ">Polar (solvatos carbocatión) prótico (ayuda a sacar el grupo de salida y lo solvata. | \ (\ mathrm {S} _ {\ mathrm {N}} 2\)” style="height:15px; ">Polar aprótico: solvata el catión dejando al nucleófilo sin solvatar y más reactivo. |
| Estereoquímica | \ (\ mathrm {S} _ {\ mathrm {N}} 1\)” style="height:15px; ">La racemización resulta del ataque a ambos lados del carbocatión plano | \ (\ mathrm {S} _ {\ mathrm {N}} 2\)” style="height:15px; ">Inversión de la configuración si la reacción tiene lugar en el centro quiral |
En la práctica, los sustratos terciarios solo sufren\(\mathrm{S}_{\mathrm{N}} 1\) y el metilo y el primario solo sufren\(\mathrm{S}_{\mathrm{N}} 2\), son los sustratos secundarios donde radica la ambigüedad, y para ello hay que considerar los otros factores como la resistencia solvente y nucleofílica. Sin embargo, las reacciones que generan carbocationes también pueden experimentar otras vías de reacción además de la sustitución.