22.3: Métodos moleculares para estudiar la organización del genoma nuclear
- Page ID
- 54938

Existen dos tipos principales de métodos para investigar la estructura tridimensional de la cromatina en el núcleo.
- El primer conjunto de métodos, ChIP y DaMid, son métodos que miden las interacciones ADN-'Landmark'. Es decir, miden interacciones de loci genómicos con hitos nucleares relativamente fijos, y solo se identificarán regiones del genoma que entren en contacto con la lámina nuclear.
El segundo conjunto de métodos, los métodos basados en 3C, son aquellos que miden las interacciones ADN-ADN. Pueden identificarse dos regiones cualesquiera de ADN que interactúen, independientemente de que estén cerca del interior o de la periferia del núcleo.
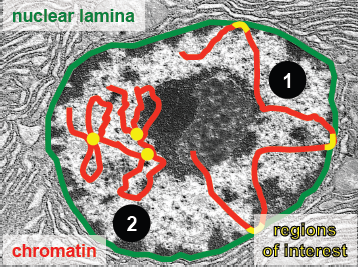
Métodos para medir las interacciones ADN-Lamina Nuclear
Los siguientes métodos, ChIP y DaMid, examinan regiones del ADN que específicamente entran en contacto con la lámina nuclear.
ChIP: Cromatina Immuno Precipitación
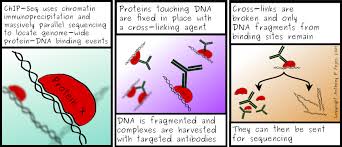
ChIP es un método para detectar regiones de ADN que están unidas a proteínas de interés. Las proteínas unidas con ADN se reticulan en su lugar con formaldehído. Los complejos proteína-ADN se extraen mediante cromatografía de anidad, principalmente utilizando anticuerpos específicos que se dirigen a la proteína de interés. Luego se desasocian los complejos recuperados, se rompen las reticulaciones y se fragmenta y analiza el ADN que se unió a las proteínas. Los fragmentos de ADN se pueden analizar usando secuenciación (ChIP-seq) o micromatrices (Chip-chip). Sin embargo, un gran desafío asociado con las diversas técnicas de ChIP es que puede ser difícil obtener un anticuerpo de alta anidad. Para estudiar la estructura 3D del ADN dentro del núcleo, se puede usar Chip-seq con anticuerpos que encaran las proteínas de la lámina.
DAMID: Identificación de ADN adenina metiltransferasa
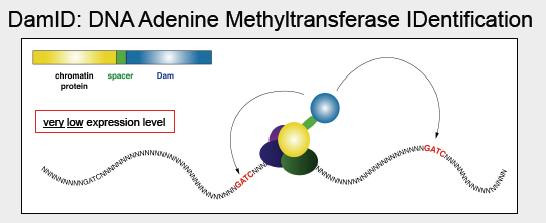
La DAMID se usa para mapear los sitios de unión de las proteínas de unión a cromatina. En el método DaMid, la ADN adenina metiltransferasa (Dam) de E. coli se fusiona a la proteína laminB1 (la enzima Dam cuelga del extremo de la proteína y, por lo tanto, está en las proximidades para interacciones). En E. coli, la enzima Dam metila la adenina en la secuencia GATC; los genomas bacterianos contienen proteínas con funciones como Dam para proteger su propio ADN de la digestión por enzimas de restricción, o como parte de sus sistemas de reparación de ADN. Como este proceso no ocurre naturalmente en eucariotas, las adeninas metiladas en una región pueden atribuirse así a una interacción con la proteína fusionada con Dam, lo que implica que esa región particular entró en contacto cercano con la lámina nuclear. Como control, la Presa sin fundir se puede expresar en niveles bajos. Esto da como resultado una distribución dispersa de adenina metilada para la cual la posición precisa de las adeninas metiladas se puede utilizar para inferir la variación en la accesibilidad del ADN. Las adeninas metiladas se determinan mediante ensayos de PCR de disulfuro u otra técnica de PCR sensible a las metilaciones en el ADN molde. En uno de esos ensayos, el genoma puede ser digerido por DpNi, que sólo corta secuencias GATC metiladas. Las secuencias adaptadoras se ligan entonces a los extremos de estas piezas digeridas, y la PCR se ejecuta usando cebadores que coinciden con los adaptadores. Solo se amplifican las regiones que ocurren entre las posiciones proximales del GATC. La medición final es el log de la relación entre la asociación de lámina de prueba y control: los valores positivos se asocian preferentemente a la lámina, y por lo tanto se identifican como LAD. Una ventaja de usar DaMid sobre ChIP es que DaMid no requiere un anticuerpo específico que puede ser difícil de encontrar. Sin embargo, una desventaja del uso de DaMid es que la proteína de fusión debe elaborarse y expresarse.
Cortesía de Anthony P. Fejes. Usado con permiso.
Cortesía de Bas van Steense. Usado con permiso.
FAQ
P: ¿Qué tan cerca tiene que llegar el ADN a la DAMID para ser metilada?
R: No tiene que unirse directamente a la lámina, pero sí tiene que acercarse bastante. El DAMID tiene un rango de aproximadamente 1.5kb.
Medición de contactos ADN-ADN
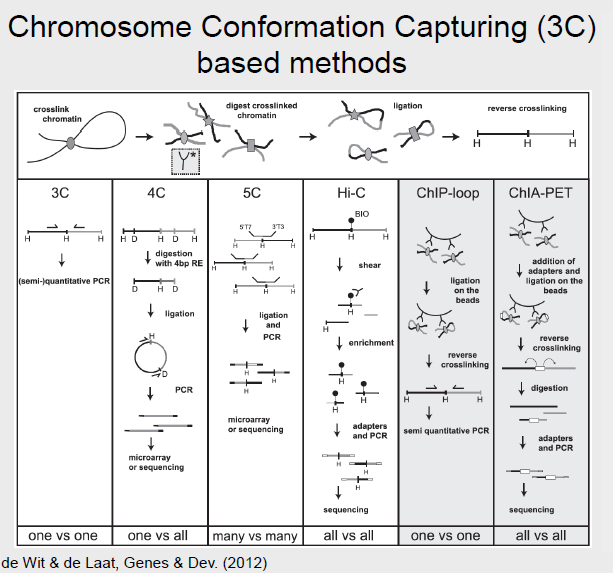
Todos los siguientes métodos se basan en la Captura de Conformación Cromosómica (3C) con ciertas modificaciones.
Prensa de Laboratorio Cold Spring Harbor. Todos los derechos reservados. Este contenido está excluido de nuestro Creativo
Licencia Commons. Para obtener más información, consulte http://ocw.mit.edu/help/faq-fair-use/.
Fuente: de Wit, Elzo y Wouter de Laat. “Una década de Tecnologías 3C: Insights into Nuclear Organization”.
Genes & Desarrollo 26, núm. 1 (2012): 11-24.
3C
La captura de conformación cromosómica (3C) es un método que detecta qué loci genómicos están cerca de otros loci dentro del núcleo. Similar al método ChIP, se utiliza un agente de reticulación para congelar proteínas unidas al ADN en su lugar y formar complejos proteína-ADN. El ADN puede entonces ser digerido por una enzima de restricción después de permitir que la proteína unida se disocie. Por lo general, se usa una enzima con un sitio de reconocimiento de 6 bps de largo que deja extremos pegajosos, como HindIII. Los fragmentos generados son inducidos entonces a autoligarse (Usando concentraciones muy bajas de ADN para evitar la ligación del fragmento con otro fragmento aleatorio). El resultado es un conjunto de fragmentos lineales de ADN, conocidos como la biblioteca 3C, que pueden analizarse mediante PCR diseñando cebadores específicamente para la interacción de interés. 3C puede describirse como un método 'uno contra uno', ya que los cebadores utilizados son específicamente diana para amplificar el producto de la interacción entre 2 regiones de interés.
Captura de conformación de cromatina circularizada (4C)
Los métodos 4C pueden describirse como 'uno contra todos' porque para una sola región de interés, podemos examinar todas sus interacciones con todas las demás regiones del genoma. 4C funciona de manera similar a 3C siendo la dierencia principal la enzima de restricción utilizada. En 4C, se emplea un cortador común para generar más y más fragmentos más pequeños. Estos fragmentos se ligan luego de nuevo. Algunos fragmentos más pequeños pueden ser excluidos, pero el resultado es un fragmento circularizado de ADN. Se pueden diseñar cebadores para amplificar el fragmento 'desconocido' de ADN de manera que se identifiquen todas las interacciones con la región de interés.
Captura de conformación cromosómica con copia de carbono (5C)
5C es un método 'muchos vs muchos' y permite la identificación de interacciones entre muchas regiones de interés y muchas otras regiones, también de interés, para ser analizadas a la vez. 5C funciona de manera similar a 3C. Sin embargo, después de obtener la biblioteca 3C, se realiza la amplificación mediada por ligación múltiple (LMA). La MLA es un método en el que se amplifican múltiples dianas. La biblioteca 5C resultante puede analizarse en una micromatriz o secuenciación de alto rendimiento.
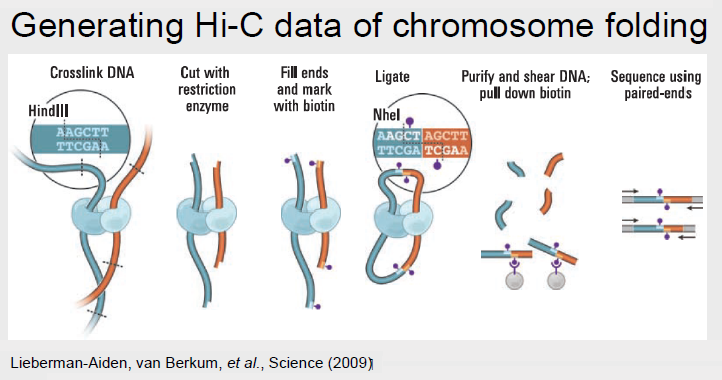
Hi-C
Hi-C puede describirse como un método 'todo contra todos' porque identifica todas las interacciones de la cromatina. Hi-C funciona marcando todos los fragmentos de ADN con biotina antes de la ligadura, lo que marca todas las uniones de ligadura. Luego se usan perlas magnéticas para purificar las uniones marcadas con biotina. Esta biblioteca Hi-C puede entonces ser alimentada en la secuenciación de próxima generación.
Chip-loop
Chip-loop se puede describir como un método 'uno contra uno', ya que similar al 3C, solo se puede identificar una interacción entre dos regiones de interés. Chip-loop es un híbrido entre ChIP y los métodos 3C. Los complejos ADN-proteína se reticulan primero y se digieren. Entonces, como en ChIP, la proteína de interés y el ADN unido a ella son arrastrados hacia abajo usando un anticuerpo. El protocolo luego procede como en 3C: los extremos libres de los fragmentos se ligan, la reticulación se invierte y la secuenciación puede proceder usando cebadores diseñados específicamente para una interacción 'uno contra uno'.
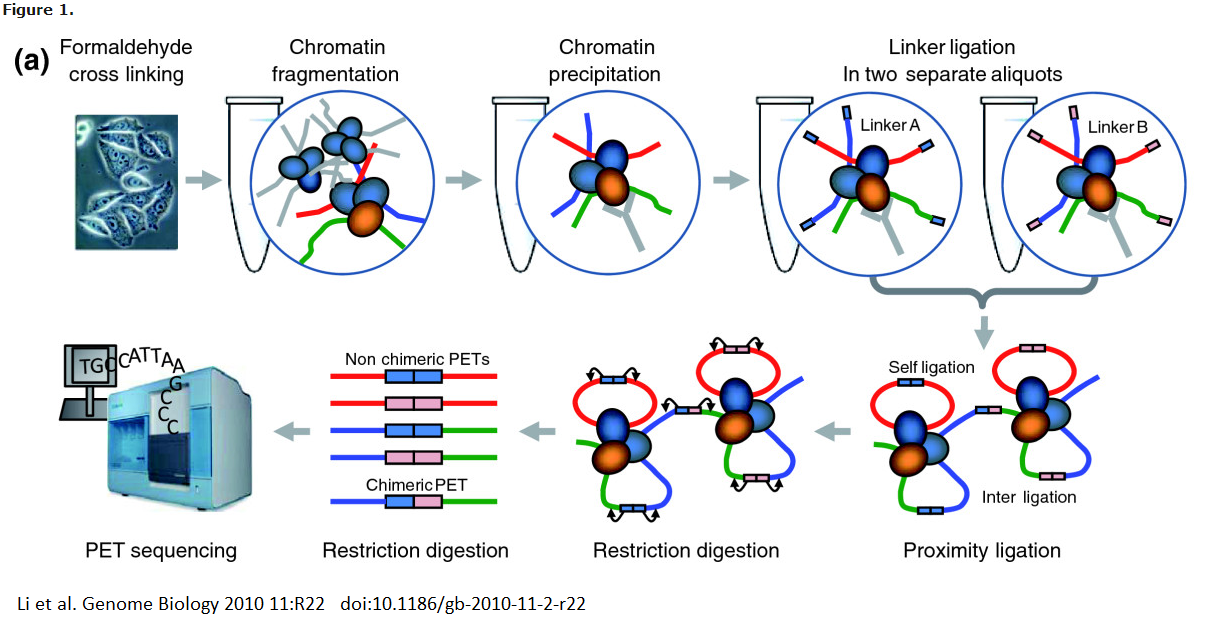
Chia-pet
Análisis de Intercepción de Cromatina por Secuenciación de Etiquetas de Extremo Pareado, o Chia-PET, combina las metodologías ChIP y 3C para determinar las interacciones de cromatina de largo alcance en todo el genoma. Se puede describir como un método 'todo vs todos', ya que aunque se debe identificar una sola proteína de interés, se identificará cualquier interacción. En Chia-PET, los complejos ADN-proteína están reticulados, como en los métodos previamente discutidos. Sin embargo, la sonicación se usa para romper la cromatina y reducir las interacciones no específicas. Al igual que en el protocolo ChIP, se usa un anticuerpo para tirar hacia abajo regiones de ADN unidas a una proteína de interés. Dos enlazadores oligonucleótidosdierentes se ligan entonces a los extremos libres del ADN. Ambos enlazadores tienen sitios de corte MMeI. Los enlazadores se ligan entonces entre sí para que los extremos libres se conecten, después de lo cual los fragmentos se digieren con MmeI. MmeI corta 20 nt aguas abajo de su secuencia de reconocimiento, por lo que el resultado de la digestión es el enlazador bordeado por la secuencia de interés en ambos lados. Esta es una estructura de 'etiqueta-enlazador-etiqueta', y los fragmentos se conocen como PET. Las PET pueden secuenciarse y mapearse de nuevo al genoma para determinar regiones de ADN que interactúa.