
22.4: Mapeo de interacciones genoma-lámina nuclear (LADs)
- Page ID
- 54926

En esta sección, presentaremos cómo se utilizaron los métodos DAMID y Hi-C para mapear dominios asociados a láminas en el genoma.
Interpretación de datos de DAMID
Se utilizó el método DAMID (descrito en la sección 3) para identificar regiones de ADN que interactuaban con la proteína de lámina en la lámina nuclear.
¿Sabías?
Los experimentos de DAMID normalmente se ejecutan durante 24 horas y la metilación es irreversible. Los resultados también son el promedio sobre millones de células. Por lo tanto, el DAMID no es adecuado para el posicionamiento del genoma relacionado con el tiempo exacto, aunque los estudios unicelulares pronto podrán hacer abordar este tema!
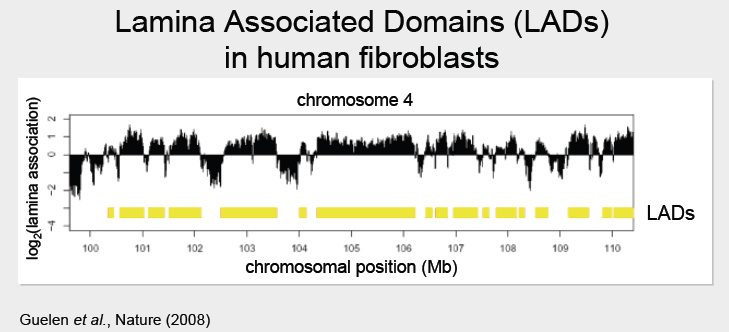
Los resultados del experimento de DAMID se graficaron como\(\log _{2} \frac{\text {Dam fusionprotein}}{\text {Dam only}}\) proteína, tal como se hizo en la siguiente figura (picos negros). Para el experimento de fusión LaminB1, las regiones positivas (subrayadas en amarillo en la figura siguiente) indican regiones que se asocian preferentemente con la lámina nuclear. Estas regiones positivas son desmultadas como Dominios Asociados a Lamina, o LADs. Aproximadamente 1300 LAD fueron descubiertos en humanos
fuente desconocida. Todos los derechos reservados. Este contenido está excluido de nuestro Creativo
Licencia Commons. Para obtener más información, consulte http://ocw.mit.edu/help/faq-fair-use/.
fibroblastos. Eran sorprendentemente grandes, oscilando entre aproximadamente 0.1Mb - 10Mb, con un tamaño mediano de 5Mb.
¿Sabías?
Los experimentos de DAMID normalmente se ejecutan durante 24 horas y la metilación es irreversible. Los resultados también son el promedio sobre millones de células. Por lo tanto, el DAMID no es adecuado para el posicionamiento del genoma relacionado con el tiempo exacto, aunque los estudios unicelulares pronto podrán hacer abordar este tema!
FAQ
P: ¿En esta representación es significativo un valor de 0?
R: No, definitivamente hay un punto donde no sabemos dónde está el 0 real. En cambio, podemos intentar hacer una buena estimación de dónde debe estar el valor 0, para ver la preferencia relativa (el interior vs exterior del núcleo)
Después de identificar los LAD, podemos alinear sus límites para descubrir diversas características interesantes como densidades génicas conocidas o niveles de expresión génica a los datos para construir nuestro modelo de LAD. Los experimentos han demostrado que los LAD se caracterizan por bajos niveles de densidad génica y expresión génica. Se notó que los límites de la LAD están muy definidos. Al alinear las posiciones de inicio de muchos LAD, se descubrió que los bordes están particularmente marcados por islas CpG, promotores que apuntan hacia afuera y sitios de unión a CTCF.
FAQ
P: ¿Por qué sitios de unión a CTCF? ¿Qué tienen de importante ellos?
R: ¡Esa es la pregunta! Quizás ayuden a mantener a los LADs. Quizás podrían impedir que los LADs se 'extendier'.
FAQ
P: ¿Cómo se relaciona la organización con la expresión policlonal?
R: Ciertamente algo sucede; sin embargo, la represión policlonal funciona a menor escala que la LAD. Ocurre fuera de los LADs, como mecanismo de represión adicional
Interpretación de datos Hi-C
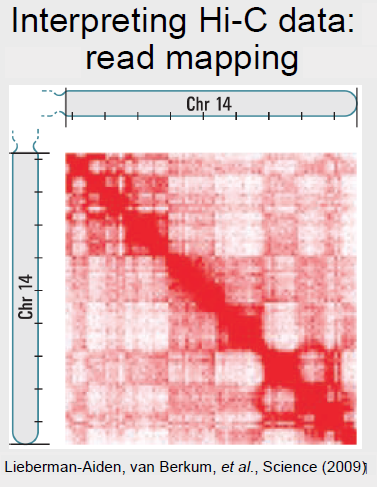
Se recolectaron datos Hi-C y la lectura se mapeó de nuevo al genoma. Los recuentos de lecturas se compilaron en una matriz O (que se muestra a continuación para el cromosoma 14) donde el elemento O i, j indica el número de lecturas correspondientes a una interacción entre las posiciones i y j. Una diagonal fuerte está claramente presente, e indica que las regiones que están cercanas entre sí en 1D también son probables para interactuar en 3D. Los errores en la interpretación de los datos Hi-C pueden ocurrir cuando se violan los supuestos de la técnica: por ejemplo, la suposición de que el genoma de referencia es correcto, lo que puede no ser cierto en el caso de una célula cancerosa. La matriz fue entonces
Asociación Americana para el Avance de la Ciencia. Todos los derechos reservados. Este contenido está excluido de nuestro Creativo
Licencia Commons. Para obtener más información, consulte http://ocw.mit.edu/help/faq-fair-use/.
Fuente: Lieberman-Aiden, Erez, et al. “El mapeo integral de interacciones de largo alcance revela principios plegables de
el Genoma Humano”. Ciencia 326, núm. 5950 (2009): 289-93.
normalizado para tener en cuenta la distancia genética entre dos regiones, y una matriz que indica qué interacciones están enriquecidas o agotadas en los datos. Para comparar los datos en la matriz, que es bidimensional, con conjuntos de datos genómicos, que son unidimensionales, se debe utilizar el Análisis de Componentes Principales (PCA). Después del PCA, es posible la caracterización funcional de los datos. Hi-C identificó dos tipos globales de regiones:
• Tipo A, que se caracteriza por cromatina abierta, riqueza génica y marcas activas de cromatina.
• Tipo B, que se caracteriza por la cromatina cerrada, y es pobre en genes.
Ambos tipos de regiones son principalmente autointeraccionantes y las interacciones entre los dos tipos son infrecuentes. Hi-C también confirmó la presencia de territorios cromosómicos, ya que hubo muchas más interacciones intracromosómicas que intercromosómicas.